|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 4321 | Gradito: |
Leggi anche appunti:Classificazione dei virusCLASSIFICAZIONE DEI VIRUS I virus sono suddivisi a seconda dell'ospite parassistato Cultura medico sanitariaCULTURA MEDICO SANITARIA La demenza di Alzheimer è una patologia cerebrale Linfoma di hodgkin lhLinfoma di Hodgkin LH Il LH è una neoplasia ad insorgenza principalmente |
 |
 |
REUMATOLOGIA PEDIATRICA
La malattia reumatologica pediatrica viene evidenziata per la presenza di dolore.

Figura : Principali cause di dolore articolare in età pediatrica.
Spesso si tratta di traumi, cause meccaniche ma è importante effettuare una attenta diagnosi differenziale.
Diagnosi: Anamnesi:
Familiare: collagenopatie, malattie intestinali (mici), malattie oculari (uveite), psoriasi (artrite psoriasica);
Pregressa: infezioni respiratorie o intestinali, uso di farmaci, altri sintomi associati, traumi;
Specifica: durata e caratteristiche del dolore, tumefazione, alterazioni del termotatto, arrossamento.
Esami Di Laboratorio Utili In Caso Di Dolore Agli Arti
Emocromo: emoglobinopatie (ad esempio l'anemia a cellule falciformi si manifesta con dolori articolari), infezioni, ARG (macrocitosi, leucopenia, piastrinopenia), LES, leucemia;
VES: infezioni, infiammazione, anche spia di un tumore, una leucemia;
PCR: infezioni, ARG;
Immunoglobuline: ipogamma, deficit Ig A spia di immunocompromissione;
TAS: malattia reumatica;
Fattore reumatoide: ARG;
ANA: ARG, LES;
Ab anti Borrelia: Malattia di Lyme;
Tampone faringeo: malattia reumatica;
Es. urine: porpora di SH per verificare presenza di ematuria, S. di Reiter, LES con presenza di screzio nefritico;
Rx: discite, infezioni, M di Perthes, fratture occulte, neoplasie;
Puntato midollare: leucemie.
L'attivazione immunitaria può essere indicata dalla presenza di immuncomplessi, Ig sieriche, antigene del fattore di Von WILLEBRAND.
Terapia: La terapia antiinfiammatoria NON occulta patologie serie come neoplasie, infezioni o artriti croniche all'esordio, quindi può essere liberamente adoperata. Il cortisone può determinare una riduzione del dolore ed ha un effetto anche sulla patomorfosi sottostante mentre gli antiinfiammatori no. Prima di arrivare alla diagnosi si possono somministrare gli antiinfiammatori.
Artrite reumatoide giovanile
Gruppo clinicamente eterogeneo di artriti croniche che si presentano in età pediatrica (<16 anni).
L'artrite reumatoide giovanile (ARG) è simile all'AR adulta. La malattia tende a colpire le grandi e piccole articolazioni e può interferire con la crescita e lo sviluppo. Si può verificare micrognazia (mento sfuggente) da crescita mandibolare alterata.
La prevalenza in età scolare varia dal 6,5% della casistica inglese allo 0,8% di quella canadese. È autoimmunitaria con produzione di autoanticorpi soprattutto verso i nuclei cellulari (ANA), comunque oggi è ragionevole pensare che le varie forme di ARG abbiano diverse origini patogenetiche con diversi fattori genetici e ambientali. Infatti ecco alcune associazioni con antigeni HLA:
forma poliarticolare: DRB1 *0801 e *1401;
forma oligoarticolare (late-onset): DRB1 *0101 e 0801;
forma oligoarticolare (early onset): A2, DRB1 0801, 1101 e 1301, DPB1 *0801, 1101 e 1301, DPB1 *0201.
Artrite sistemica o morbo di Still
Si verifica in circa l'11-14% dei pazienti. Spesso compaiono febbre elevata per almeno 2 settimane, rash cutaneo maculo-papuloso transitorio, epatosplenomegalia, adenopatia diffusa, sierosite, marcata leucocitosi neutrofila e trombocitosi. Queste manifestazioni precedono a volte la comparsa dell'artrite. Il fattore reumatoide e gli anticorpi anti-nucleo sono assenti. VES e PCR elevate, piastrinosi e leucocitosi.
Prognosi: prosegue nel giro di qualche anno come forma poliartritica; può essere riacutizzata da una flogosi delle alte vie (streptococco, rhinovirus, herpes); a distanza di 10-15 anni il 25 % circa dei soggetti ha una disabilità moderata-severa o addirittura morte. Fattori di rischio per un'evoluzione cattiva sono la persistenza della febbre o la necessità di corticosteroidi a 6 mesi dall'inizio e la piastrinosi. Senza terapia una complicanza è la sindrome emofagocitica (può essere mortale): terapia: cortisonici i.v.+ ciclosporina.
Terapia: FANS ma più spesso steroidi per periodi di qualche settimana; nei casi refrattari metothrexate.
Oligoartrite persistente early onset
Colpisce 1-4 articolazioni (polsi, ginocchia, anche, piedi) e spesso coesiste uveite; frequentemente gli ANA sono presenti. Colpisce più frequentemente le femmine con picco a 3 anni.
Se ne distinguono due tipi:
TIPO I (artrite cronica giovanile): esordisce nei primi 5 aa di vita, è prevalente nel sesso femminile, mostra una positività agli ANA nel 90% dei casi e si associa al rischio di iridociclite cronica;
TIPO II: esordisce in età scolare con prevalenza nel sesso maschile (come spondiloartropatia?). Vi è la presenza di entesopatie e negatività del FR e degli ANA. Si riscontra HLA B27 +.
Prognosi: buona, potendo andare in remissione in 4-5 anni. La ripresa funzionale dipende dal trattamento.
Terapia: spesso necessita cortisonici intra-articolari.
Oligoartrite estende o late onset
Inizia come oligoartrite ma nel giro di un anno evolve in multiartrite.
Prognosi: buona, potendo andare in remissione in 4-5 anni. La ripresa funzionale dipende dal trattamento.
Terapia: risente favorevolmente della tp con metotrexate.
Poliartrite
Sono forme quasi sempre siero-negative, simili a quelle dell'adulto. Un sottogruppo è stato individuato di recente caratterizzato da rigidità e contratture muscolari. Si manifesta nel teenager.
Di questo quadro clinico se ne riscontrano due forme:
FORMA FR+ (sieropositiva): si caratterizza oltre che per la presenza del FR molto aumentato anche per la presenza di un HLA DR4, ANA positivo nel 40-80% dei casi. In questa forma sono principalmente interessate le piccole articolazioni;
FORMA FR- (sieronegativa): oltre al FR negativo si ritrovano ANA positivi. Il quadro clinico è eterogeneo ma più frequentemente si caratterizza per un esordio oligoarticolare con interessamento delle grandi articolazioni.
Prognosi: cattiva, con ampie distruzioni articolari. Possono arrivare nell'adulto alla distruzione dell'articolazione con necessità di rimpiazzo di questa a 20-30 anni.
Terapia: FANS e methotrexate (mg 10-20/m2/sett. os); quest'ultimo nei casi resistenti si può usare anche forti dosi.
Enthesitis arthritis
È una forma caratterizzata da artrite asimmetrica delle articolazioni dell'arto inferiore ed entesite (flogosi della fascia muscolare), che colpisce i bambini più grandi ed è associata a HLA B27 e uveite (forse è l'equivalente della spondilite anchilosante verso cui evolve nel 60 % dei casi dopo 15 anni); la sacroileite molto di rado si trova in età pediatrica.
Prognosi: non c'è evoluzione verso una artrite erosiva né verso una grave compromissione funzionale.
Terapia: sulphalazina (2-3 g/Kg)+ FANS (indometacina). Nel caso frequente di evoluzione verso una forma sistemica è indicato il methotrexate.
Artrite psoriasica
 È caratterizzata da infiammazione delle dita
delle mani e dei piedi all'interno di una psoriasi in età pediatrica. Può
comunque colpire anche le articolazioni grandi e piccole (poliartrite) fino a
quadri altamente erosivi.
È caratterizzata da infiammazione delle dita
delle mani e dei piedi all'interno di una psoriasi in età pediatrica. Può
comunque colpire anche le articolazioni grandi e piccole (poliartrite) fino a
quadri altamente erosivi.
Terapia: difficile (methotrexate?).
![]() Terapia
Terapia
Il trattamento di tutte le forme si deve associare ad un approccio riabilitativo e di supporto psico-sociale. I farmaci si suddividono in tre classi:
FANS;
CORTISONICI;
FARMACI DI SECONDA LINEA o modificanti la malattia: sono farmaci da usare dopo il fallimento dei primi due gruppi ed in grado di modificare l'andamento della malattia.
Methotrexate: agisce a diversi livelli: sopprime l'immunità cellulo-mediata, riduce la concentrazione di interleukina 1β nel liquido sinoviale ed ha un effetto antiproliferativo sui fibroblasti della sinovia.
Ciclosporina: agisce bloccando le cellule T e quindi inducendo una riduzione dell'interleukina-2. È molto nefrotossica.
Sulfalazina (combinazione di salicilato e sulfapiridina).
Azatioprina (Imuran).
Agenti alchilanti (es. ciclofosfamide o clorambucil): pochissimo usati per i loro maggiori effetti tossici (anche cancerogeni: linfomi e tumori della vescica) e teratogeni.
Sali d'oro: ci sono prodotti sia i.m. sia orali (Auranofin); possono provocare piastrinopenia, lesioni renali, reazioni allergiche cutanee anche gravi.
Anti-malarici (idrossi-clorochina).
Infliximab (Remicade) è un anticorpo chimerico, umano-murino, monoclonale, che si lega con alta affinità sia alla forma solubile, che a quella transmembrana del TNF-alfa (Tumor Necrosis Factor). Va somministrato insieme al methotrexate. Si è dimostrato che nel tempo si possono formare anticorpi bloccanti l'azione dell'infliximab: la contemporanea presenza del methotrexate rallenta la formazione di detti anticorpi. L'infliximab è disponibile solo per via endovenosa, ragion per cui è somministrato in Ospedale per fleboclisi. Dopo una prima somministrazione ne segue una seconda dopo due settimane e una terza dopo altre quattro settimane; successivamente le infusioni sono effettuate ogni otto settimane.
Etanercept: è un farmaco anti-TNF-alpha che fin dalle prime fasi sperimentali si è dimostrato attivo nelle forme d'artrite refrattaria alle altre terapie. Può essere usato da solo o in combinazione con il methotrexate. Gli studi effettuati sulle artriti esordite da meno di tre anni hanno dimostrato che il farmaco è attivo almeno tanto quanto il methotrexate nel migliorare i sintomi e nel rallentare la progressione radiologica delle erosioni. È generalmente ben tollerato ed è somministrato sottocute due volte la settimana (anche dal paziente stesso). Il blocco del TNF riduce si l'infiammazione, ma anche la capacità di combattere le infezioni da parte del nostro sistema immunitario (facilità alla TBC).
Anakinra: appena uscito dalla fase sperimentale; agisce come anti-interleukina 1, una citochina proinfiammatoria che si produce nei tessuti infiammati. È prodotto mediante la tecnologia del DNA ricombinante utilizzando Escherichia coli come sistema di espressione; si somministra sottocute, una volta al giorno, specie associato al Methotrexate (una volta la settimana).
Tutti questi farmaci di secondo livello sono in grado di dare un miglioramento dei sintomi dolorosi e dell'indice di disabilità.
Un altro parametro da tener conto per valutare l'efficacia della terapia è l'aspetto radiologico (mediante Rx standard, TAC e RMN) dell'articolazione colpita (comparsa di nuove erosioni e vari tipi di punteggi radiologici) e l'andamento degli indici infiammatori (VES, proteina C).
Malgrado ciò numerosi studi (anche in meta-analisi) hanno evidenziato che non esiste un farmaco sicuramente superiore ad un altro, soprattutto a breve termine (< 1 anno).
L'uso dei vari farmaci di 2° livello è ancor oggi senza chiare linee guida ed è del tutto empirico. Essi vanno riservati ai pazienti che non trovano beneficio dagli antinfiammatori (FANS e cortisonici). In effetti la maggior parte dei pazienti deve ricorrere a questi farmaci.
Secondo un numero sempre più vasto di clinici questi farmaci vanno usati sempre più precocemente, specie nei pazienti con grave sinovite o che abbiano fattori di rischio di cattiva prognosi (positività del FR, noduli sottocutanei o altri segni extra-articolari, gravi segni radiologici).
In genere i farmaci di 2° livello sono usati in associazione ai FANS e cortisonici (effetto sinergico) e l'effetto va valutato sul piano clinico: valutazione del dolore, valutazione clinica dello stato infiammatorio dell' articolazione colpita, stato di disabilità ed eventualmente anche con gli indici infiammatori (i dati immunologici e il reperto radiografico non sono utili per valutare l'efficacia della terapia).
Il periodo di osservazione prima di fare modifiche nei farmaci e nei dosaggi deve essere di alcuni mesi (4-6 mesi).
Il trattamento viene intrapreso in base al sottotipo di malattia, alla sua gravità, alle manifestazioni specifiche e alla risposta alla terapia.
Primo livello: FANS (terapia sintomatica: controllo del dolore o nelle forme sistemiche della febbre) + fisioterapia. I vari FANS non sono equivalenti ma presentano vantaggi e svantaggi che vanno tenuti presenti in particolar modo in ambito pediatrico:
ACIDO ACETILSALICILICO (ASA): 60 -120 mg/Kg/die/4dO (poco usato: epatotossicità importante, dosaggio almeno 4dO/die) . Si consiglia la somministrazione dopo i 12 anni;
NAPROSSENE: 15 - 20 mg/Kg/die/2dO (molto usato nelle forme poli e oligo, pochi effetti collaterali);
IBUPROFENE: 30 - 40 mg/Kg/die/3dO;
FLURBIPROFENE 5 - 7,5 mg/Kg/die/3dO;
INDOMETACINA: 3 - 5 mg/Kg/die/3dO (ottima azione antipiretica nelle forme sistemiche, associato al naprossene quando vi è spiccata rigidità mattutina utilizzando metà della dose/die in unica somministazione serale per via rettale);
TOLMETINA: 20 - 30 mg/Kg/die/3-4 dO;
PIROXICAM: 5 - 20 mg/Kg/die/1dO.
La risposta individuale ai FANS è estremamente variabile e va valutata dopo un periodo di almeno 4-8 settimane.
La terapia gastroprotettiva (anti H2) è raramente necessaria. La terapia steroidea (prednisone 1-2 mg/Kg/die) è riservata alle forme sistemiche con imponente sintomatologia generale (febbre, pericardite) o alle forme oligoarticolari con grave uveite.
Secondo livello: FANS + Methotrexate + fisioterapia e tutorazione. Il Methotrexate:è dato a dosi di 10 mg/m2, e si richiede il controllo di funzionalità ed ecografia epatica (epatotossicità e fibrosi epatica), funzionalità polmonare, funzionalità renale e midollare.
Possono essere effettuati dei cicli se è necessario continuare la terapia per lungo tempo. Si procede con l'aumento della dose di methotrexate da 10 mg/m2 a 30 mg/m2 ,poi si aggiunge la ciclosporina.
Terzo livello: FANS + Methotrexate ad alte dosi o immunosoppressori + fisioterapia. Methotrexate: 30 mg/m2 oppure Methotrexate: 10 mg/m2 + ciclosporina. Somministrando il methotrexate si rischia la sterilità però se non trattato rischia problemi di mobilità.
Quarto livello: FANS + Ab monoclonali anti TNF-α + fisioterapia e chirurgia degli esiti.
Etanercept (Enbrel): approvato per l'uso nell'ARG; 5 mg/Kg/sc 2 volte a settimana;
Infliximab (Remicade): approvato per M. di Crohn e AR non per ARG, solo ospedaliero; 5 mg/Kg/die EV a T0, T15, T45 poi ogni 2 mesi. Riportata comparsa di autoAb e rischio di infezioni gravi, maggiore efficacia con MTX 10 mg/m2.
Alcuni centri specialistici saltano il 2^ e 3^ livello di terapia e si inizia un protocollo sperimentale con gli Ab monoclonali che comunque possono dare un quadro immunosoppressivo e complicanze di tipo neoplastico a lungo tempo..
Prognosi
Pochi pazienti hanno una completa remissione della malattia; pochi hanno una evoluzione resistente a tutti i farmaci; la maggior parte trova prima o poi beneficio da uno dei farmaci di 2° livello.
In genere la sospensione del farmaco fa ritornare i sintomi, pertanto la terapia va sospesa solo in casi molto particolari (benessere per lungo tempo oppure comparsa di effetti secondari o tossici).
Uno studio USA mostra che il 50 % dei soggetti ha la malattia attiva dopo 15 anni. Come tutte le malattie croniche infiammatorie provoca:
ritardo di crescita generale e accelerata crescita a livello locale (epifisi delle articolazioni colpite);
ridotta frequenza a scuola;
ridotta possibilità di lavoro: l'impatto sociale della malattia dipende molto dall'indice di disabilità calcolato su appositi questionari: CHAQ (Childhood Health Assessment Questionnaire). I valori più alti si riscontrano nelle forme poliarticolari e nei soggetti con FR+ (IgM).
Complicanze dell'ARG
Ritardo di crescita e alterazione dello stato nutrizionale se si utilizzano steroidi per lungo tempo;
Osteoporosi;
Amiloidosi;
Iridociclite cronica.
Si possono verificare complicanze ortopediche che comprendono una differente lunghezza degli arti inferiori, cisti poplitee e contratture da flessione.
Ritardo di crescita: Il ritardo di crescita può essere segmentario o più frequentemente generalizzato. A sua volta quest'ultimo si può distinguere in una forma sistemica o in una forma poliarticolare. Fattori che condizionano negativamente la crescita sono:
Attività della malattia;
Durata;
Precocità di esordio (i bambini restano nani);
terapia steroidea.
Riduzione della massa ossea: Quando parliamo di riduzione della massa ossea distinguiamo due condizioni a seconda della quantità di sostanza perduta ed in particolare se la perdita è stata di 1 - 2 SDS si parla di osteopenia, se invece la perdita è stata > 2 SDS si parla di osteoporosi. L'osteoporosi a sua volta può essere localizzata a livello iuxtaarticolare o essere generalizzata.
Ci sono alcuni fattori che concorrono alla perdita della massa ossea nei pazienti con ACG e distinguiamo fattori più comuni e generici, non correlati strettamente alla patologia e fattori specifici:
Generali: Sesso femminile, razza bianca, ridotto apporto di Ca, ridotta esposizione solare, limitata attività fisica;
Specifici: ormoni, citochine infiammatorie IL 1 , IL 6 , TNF-α, terapia steroidea. Per quanto riguarda gli steroidi la loro azione a livello del metabolismo osseo è notevole ed importante. Essi riducono l'assorbimento di Ca interstiziale, aumentano la quota di Ca e P urinario che viene perso, riducono l'attività osteoblastica, la differenziazione degli osteoclasti ed i recettori della vit. D, mentre potenziano l'attività osteoclastica (iperparatiroidismo).
Metodiche per il rilievo dell'osteoporosi:
Radiografia: scarsa sensibilità;
Tomografia QCT;
Assorbimetria a fotone singolo (SPA);
Assorbimetria a fotone doppio (DPA);
Assorbimetria con raggi X a doppia energia (DEXA);
Attenuazione degli ultrasuoni da parte dell'osso (BUA) ,poco invasiva e sensibile per l'osteopenia.
Prevenzione:
Rapida mobilizzazione;
Adeguato apporto nutrizionale;
Controllo dell'intake di Ca;
Integrazione con vit. D;
Steroidi solo nei casi più gravi;
Steroidi a dosi più basse possibili;
Riduzione dell'attività della malattia.
Prospettive terapeutiche: le prospettive terapeutiche al momento sono scarse e necessitano di ulteriori approfondimenti. L'utilizzo di Ca e vit.D ha fornito dati poco confortanti, mentre si ha scarsa esperienza in età pediatrica riguardo l'uso di Calcitonina e fluoruri. Non ci sono ancora dati conclusi per ciò che concerne i bifosfanati(aledronato).
Amiloidosi: Proteinuria e dimostrazione istologica dell'amiloide alla biopsia rettale e/o renale con prevalenza del 7 - 8 %. Intervallo tra esordio della malattia articolare sistemica e amiloide: 2 - 9 anni. Diagnosi: Amiloide (rosso congo), marcatori con radioisotopi, agobiopsia del grasso sottocutaneo.
Spondiloartropatie pediatriche (SpA)
Le SpA sono un gruppo di condizioni caratterizzate da infiammazione delle articolazioni dello scheletro assiale e/o periferico e delle entesi. Alle nostre latitudini molte SpA sono HLA-B27 negative, per cui la diagnosi non è sempre facile. Sono caratterizzate da un aumento della VES, lieve leucocitosi e piastrinosi, FR negativo. Gli ANA sono negative tranne che nel 50% dei bambini affetti da artrite psoriasica .
Tra le Spa distinguiamo:
Spondilite anchilosante giovanile;
Artriti reattive;
Artrite psoriasica;
Artriti associate a IBD;
Spondiloartriti indifferenziate.
 Criteri di classificazione delle SpA (European
SpA Study Group): Per fare diagnosi di
SpA vi deve essere sempre la presenza di dolore vertebrale infiammatorio o sinovite,
asimmetrica prevalente agli arti inferiori, accompagnati da uno dei seguenti
(sens. 77%, spec.89%):
Criteri di classificazione delle SpA (European
SpA Study Group): Per fare diagnosi di
SpA vi deve essere sempre la presenza di dolore vertebrale infiammatorio o sinovite,
asimmetrica prevalente agli arti inferiori, accompagnati da uno dei seguenti
(sens. 77%, spec.89%):
familiarità per SpA;
psoriasi;
IBD;
dolore gluteo alternante (o anche detto sciatica mozza, va in diagnosi differenziale con la sciatalgia);
entesopatia.
Se vi si ritrova la presenza di sacroileite la sensibilità diagnostica è del 86% mentre la specificità del 87%.

Sindrome Di Reiter
L'artrite è in genere oligoarticolare, si presenta con notevole gonfiore ed eritema.
Artrite psoriasica
 Presenta un interessamento articolare limitato
con distribuzione inconsueta e asimmetrica (es. piccole articolazioni della
mano e della caviglia) prima della comparsa di lesioni cutanee. E' difficile
fare diagnosi in quanto non sempre c'è la psoriasi o è presente un quadro molto
lieve di psoriasi.
Presenta un interessamento articolare limitato
con distribuzione inconsueta e asimmetrica (es. piccole articolazioni della
mano e della caviglia) prima della comparsa di lesioni cutanee. E' difficile
fare diagnosi in quanto non sempre c'è la psoriasi o è presente un quadro molto
lieve di psoriasi.
Artrite Associate a Ibd
Le malattie infiammatorie intestinali si manifestano con oligoartrite e con anemia non speigata. Interessano articolazioni assiali e periferiche. Colpiscono il 7,5 - 20% dei pz con IBD. Rappresentano il 10% di tutte le SpA giovanili. Spesso sono associate a HLA B27.


L'infezione esterna coinvolge l'articolazione mediante immunocomplessi contenenti componenti non vitali del microrganismo scatenante l'infezione. La VES risulta elevata mentre febbre e leucocitosi sono assenti.
Iperlassita' Legamentosa
Può essere causa di dolore articolare, ma anche di artrite (meccanica, secondaria al cattivo uso delle articolazioni).
 Diagnosi
Diagnosi
il pollice in abduzione tocca l'avambraccio;
le dita in iperestensione sono parallele all'avambraccio;
gomiti e ginocchia si iperestendono oltre 10°;
![]() capacità di appoggiare le mani sul pavimento con
le ginocchia in iperestensione.
capacità di appoggiare le mani sul pavimento con
le ginocchia in iperestensione.

LES
Il LES è caratterizzato da autoanticorpi diretti contro auto antigeni in grado di causare infiammazione in molti organi bersaglio compresi articolazioni, reni, cellule del sangue e SNC. Le eruzioni cutanee possono essere fotosensibili ed estendersi a tutte le zone esposte al sole. Oltre al rash malare possono comparire eruzioni maculari eritematose di tipo vasculitico, porpora e livedo reticularis e il fenomeno di Raynaud. Sono presenti anche reperti muscolo scheletrici e la sierosite che può colpire le mucose pleuriche, pericardiche e peritoneali. Manifestazioni GI si presentano con dolore, diarrea, melena, infarto, malattia infiammatoria dell'intestino ed epatite. Sono inoltre presenti manifestazioni cardiopolmonari, renali e neurologiche (SNC e SNP).
 Esami di laboratorio nel les
Esami di laboratorio nel les
Emocromo con formula: si possono rilevare anemia emolitica, leucopenia, linfopenia, piastrinopenia. Per approfondimento si possono ricercare gli ab antipiastrine e gli ab antieritrociti;
Test Di Coombs Diretto E Indiretto: Rileva la presenza di Ab liberi o adesi alla superficie dei vari elementi figurati del sangue;
![]() VES, QPE (quadro proteico elettroforetico) e
proteine totali PCR: indicativi della fase acuta dell'infiammazione, sono molto
aspecifiche;
VES, QPE (quadro proteico elettroforetico) e
proteine totali PCR: indicativi della fase acuta dell'infiammazione, sono molto
aspecifiche;
Ig: molto elevate;
C3,C4: di solito diminuiti nel plasma;
Esami Per La Funzionalita' Renale: Es. urine: creatininemia, azotemia proteinuria 24h. Se patologici si procede con un approfondimento: clearance della creatinina e poi BIOPSIA RENALE (utile per la diagnosi di nefrite lupica);
Altri esami: transaminasi (possono essere aumentate, si parla di epatite lupoide, è un'epatite autoimmune di tipo sindrome da overlap con aumentata cellularità degli spazi portali e mancata continuità della lamina limitante); gammaGT; LDH; CPK; il tutto si può approfondire con la ricerca di ISOENZIMI e ci danno conferma di un eventuale interessamento muscolare;
ECOCARDIO: versamento e/o endocardite;
DOPPLER VENOSO: Trombosi;
RX TORACE;
Prove Di Funzionalità Respiratoria;
Coinvolgimento Neuropsichico: test psicometrici per un eventuale psicosi;
tac, rachicentesi, eeg per approfondimento.
Terapia: La terapia dipende dagli organi bersaglio colpiti. La terapia steroidea ha migliorato la nefropatia e la percentuale di sopravvivenza. I corticosteroidi possono essere somministrati a giorni alterni per evitare gli effetti collaterali. Il pronto riconoscimento della malattia e il trattamento precoce sono indispensabili per gli esiti del paziente. Il LES è una malattia cronica e i pazienti devono essere monitorati a vita.
Dermatomiosite e polimiosite
La dermatomiosite giovanile è tra le più comuni miopatie infiammatorie pediatriche ed è caratterizzata da eruzione cutanea e da una debolezza muscolare simmetrica prossimale che spesso risponde alla terapia immunosoppressiva.
L'eruzione cutanea spesso inizia nelle zone esposte al sole e si manifesta come primo sintomo nel 50% dei casi e si associa a debolezza nel 25% dei casi. E' caratteristico l'eritema violaceo periorbitale (eliotropo) che può essere localizzato sulla radice del naso come una mascherina e può raggiungere anche le orecchie. La gravità dell'eritema è causata da un diminuito numero di anse capillari a livello del solco ungueale che dimostra una vascolopatia sistemica.
L'anticorpo più comunemente associato alla miosite è l'antigene polimiosite/sclerodermia (Pm/Scl).
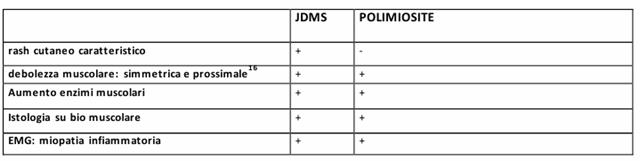
Figura : Criteri diagnostici per la polimiosite e la dermatomiosite.
PER LA DIAGNOSI: presenza (o assenza) di rash cutaneo ed esclusione di altre patologie reumatologiche + 3 criteri.

LA VES risulta normale, il FR negativo e gli enzimi muscolo derivati presentano elevati livelli nel siero. Deve essere tenuta sottocontrollo la progressione del coinvolgimento muscolare mediante esami obiettivi ed occorre eseguire esami di laboratorio per valutare l'aumento dei madiatori immuni e degli enzimi muscolari nel siero. La terapia consiste nella somministrazione di prednisone per os mentre nei pazienti con estesa vascolopatia si somministra il metilprednisone per ev.
Porpora di Henoch-Schonlein
Tale quadro morboso venne descritto per la prima volta, contemporaneamente, da Henoch e Schonlein. La PHS è una vasculite dei piccoli vasi, ANCA negativa come le crioglobulinemie, dalla durata di circa 3-4 settimane. L'incidenza è di circa 8-22 casi pediatrici ogni 100000 bambini per anno. Infatti tale quadro clinico è tipico dell'età pediatrica. In merito all'incidenza, esistono tutta una serie di lavori, presentanti valori discordanti, con oscillazioni anche ampie. Questo è forse dovuto anche all'accuratezza con la quale i singoli presidi ospedalieri effettuano diagnosi di PHS. L'età che risulta essere maggiormente colpita è quella infantile, tra i 3-15 anni, ma può colpire anche gli adulti, con una preferenza per il sesso maschile.
La PHS o porpora anafilattoide è la porpora più frequente in età pediatrica e il distacco rispetto alle altre forme è notevole, tuttavia, può colpire anche gli individui adulti. Ma negli adulti, al contrario dei bambini, la PHS si presenta con una maggiore gravità e con una maggiore probabilità di recidiva. Per quanto riguarda l'eziologia, questa risulta essere incerta. Alcuni studi hanno posto l'accento su alcune correlazioni, infatti spesso la porpora insorge a seguito di una infezione delle alte vie respiratorie, anche di natura streptococcica, e questo giustifica anche la stagionalità di tale fenomeno. In merito ad analisi epidemiologiche, si è visto che la razza caucasica risulta essere quella maggiormente colpita, e alcuni studi riportano una quantità di casi nei soggetti di tale etnia superiore al 90% dei casi totali. In merito alla stagionalità, il picco si osserva tra l'autunno e l'inverno, periodo di picco anche per le infezioni delle vie respiratorie.
Da un punto di vista clinico, la PHS si caratterizza per: porpora palpabile non trombocitopenica, coinvolgimento articolare e dolore addominale. Questa triade rappresenta la forma classica della malattia. Nel momento in cui, accanto a questa triade sintomatologica, si associa anche un coinvolgimento renale, si parla di forma complicata.
 Adesso andiamo ad analizzare i singoli elementi.
La porpora palpabile non trombocitopenica è il sine qua non della PHS. È un
esantema ed è presente nel 100% dei piccoli pazienti ed è causata da un
processo infiammatorio dei piccoli vasi. Infatti non bisogna dimenticare che si
tratta sempre di una vasculite e tutte le manifestazioni cliniche sono dovute
al processo infiammatorio localizzato a livello dei piccoli vasi. I piccoli
vasi, soprattutto quelli cutanei, vanno incontro a rottura cui segue lo
stravaso di sangue. La porpora deve essere palpabile e non è trobocitopenica,
ovvero non è dovuta alla mancata produzione, o eccessivo consumo, di piastrine.
La prima diagnosi differenziale, deve essere posta infatti con tutte le altre
forme di porpora di natura trombocitopenica. La porpora è palpabile perchè è
dovuta alla rottura dei piccoli vasi, con la formazione di un infiltrato che
evolve verso, porpora, petecchia fino ad arrivare a forme ulcerate. Alla
digito-pressione o alla vitro-pressione, la lesione non scompare ma persiste.
Compaiono gittate successive di porpora che durano da 3 a 10 giorni. Può
comparire e scomparire anche per un anno dopo la PHS. Per quanto riguarda le
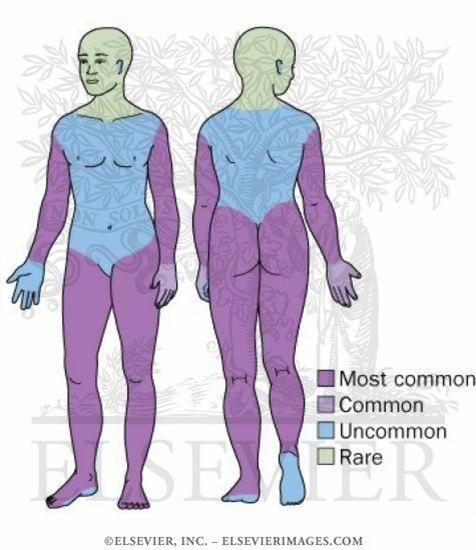
aree cutanee maggiormente colpite, queste sono schematizzate nell'immagine.
Adesso andiamo ad analizzare i singoli elementi.
La porpora palpabile non trombocitopenica è il sine qua non della PHS. È un
esantema ed è presente nel 100% dei piccoli pazienti ed è causata da un
processo infiammatorio dei piccoli vasi. Infatti non bisogna dimenticare che si
tratta sempre di una vasculite e tutte le manifestazioni cliniche sono dovute
al processo infiammatorio localizzato a livello dei piccoli vasi. I piccoli
vasi, soprattutto quelli cutanei, vanno incontro a rottura cui segue lo
stravaso di sangue. La porpora deve essere palpabile e non è trobocitopenica,
ovvero non è dovuta alla mancata produzione, o eccessivo consumo, di piastrine.
La prima diagnosi differenziale, deve essere posta infatti con tutte le altre
forme di porpora di natura trombocitopenica. La porpora è palpabile perchè è
dovuta alla rottura dei piccoli vasi, con la formazione di un infiltrato che
evolve verso, porpora, petecchia fino ad arrivare a forme ulcerate. Alla
digito-pressione o alla vitro-pressione, la lesione non scompare ma persiste.
Compaiono gittate successive di porpora che durano da 3 a 10 giorni. Può
comparire e scomparire anche per un anno dopo la PHS. Per quanto riguarda le
aree cutanee maggiormente colpite, queste sono schematizzate nell'immagine.
La porpora può comparire praticamente ovunque, ma con maggior frequenza tronco e arti, soprattutto i glutei. Collo e testa non sono mai coinvolti, anzi è una vera e propria rarità. Comunque, gli arti inferiori rappresentano l'area di maggiore interessamento.
![]() Per quanto riguarda il coinvolgimento
articolare, le sedi in parte rispecchiano quelle cutanee. Tale sintomo è
presente in circa l'80% dei soggetti ed è anche questo dovuto al processo
infiammatorio dei piccoli vasi articolari. L'insorgenza è acuta e la durata è
di alcuni giorni. La sintomatologia si risolve spontaneamente. Teoricamente
possono essere colpite tutte le articolazioni, ma in misura maggiore le
articolazioni degli arti inferiori, soprattutto ginocchia e caviglie. Il fatto
che la frequenza sia inferiore rispetto alla porpora vuol dire che si possono
trovare bambini che, pur manifestando la PHS, non presentano l'interessamento
articolare. Il dolore articolare si associa anche alla tumefazione articolare
(da non confondere con l'edema periferico che si accompagna all'eruzione
cutanea).
Per quanto riguarda il coinvolgimento
articolare, le sedi in parte rispecchiano quelle cutanee. Tale sintomo è
presente in circa l'80% dei soggetti ed è anche questo dovuto al processo
infiammatorio dei piccoli vasi articolari. L'insorgenza è acuta e la durata è
di alcuni giorni. La sintomatologia si risolve spontaneamente. Teoricamente
possono essere colpite tutte le articolazioni, ma in misura maggiore le
articolazioni degli arti inferiori, soprattutto ginocchia e caviglie. Il fatto
che la frequenza sia inferiore rispetto alla porpora vuol dire che si possono
trovare bambini che, pur manifestando la PHS, non presentano l'interessamento
articolare. Il dolore articolare si associa anche alla tumefazione articolare
(da non confondere con l'edema periferico che si accompagna all'eruzione
cutanea).
Infine il terzo elemento è dato dal coinvolgimento intestinale che è causa della sintomatologia dolorosa a livello addominale o angina intestinale. Tale sintomo è presente nel 50% dei casi è la causa è da ricercarsi sempre nell'infiammazione dei piccoli vasi gastro-intestinali. Il processo infiammatorio determina ischemia intestinale, che è la causa del dolore, che poi si accompagna ad altre manifestazioni sistemiche come nausea, vomito, diarrea ematica. Talvolta l'ischemia può essere anche talmente tanto vasta da causare una enteropatia protido-disperdente e costipazione. Molto importante sono l'invaginazione e la perforazione intestinale. Qualora si verifichino questi due eventi, il trattamento è chirurgico, poiché il bambino rischia la vita. L'insorgenza dei dolori è acuta, crampiforme nella prima settimana dalla comparsa dell'esantema e può precedere l'esordio dell'esantema.
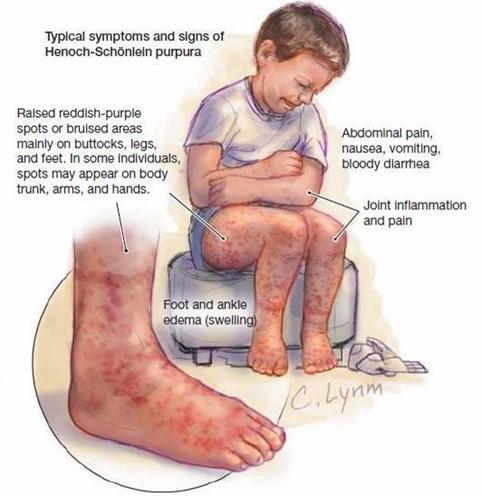 L'immagine a lato riassume quanto detto fino ad
ora, il bambino ha un atteggiamento sofferente, con le braccia cinge la pancia,
e sono presenti la porpora e il gonfiore articolare.
L'immagine a lato riassume quanto detto fino ad
ora, il bambino ha un atteggiamento sofferente, con le braccia cinge la pancia,
e sono presenti la porpora e il gonfiore articolare.
![]() Infine c'è il coinvolgimento renale. Nel momento
in cui insorge una glomerulonefrite, che può essere più o meno grave,
automaticamente si passa alla forma complicata, in quanto il rischio di morte è
maggiore così come anche delle recidive. È presente nel 30-40% dei pazienti,
ovvero 1/3 o più dei bambini sviluppa il coinvolgimento renale. Anche qui la
causa è l'infiammazione dei vasi glomerulari. L'insorgenza è acuta e la durata
variabile, anche in funzione della gravità. Nella maggioranza dei casi la
glomerulonefrite è lieve con proteinuria, ematuria microscopica e cilindri
eritrocitari. In questo caso la risoluzione è spontanea. In alcuni casi la
glomerulonefrite è importante, con proteinuria notevole, ematuria macroscopica
ed ipertensione. Questa forma è causa di una insufficienza renale acuta. Nella maggioranza
dei casi la glomerulonefrite compare nei primi mesi dalla presentazione della
PHS. In alcuni casi l'interessamento renale è tardivo e si configura come un
fattore prognostico negativo in quanto predispone ad una malattia renale
cronica. A questo punto ci si potrebbe chiedere se sia possibile prevedere
quale soggetto possa sviluppare una malattia renale cronica e stabilirne anche
la gravità. Sicuramente la proteinuria e l'ematuria sono parametri da ricercare
attraverso l'esame delle urine. Così come anche bisogna valutare bene la
funzione renale, attraverso l'eGFR. Questa tabella riassume quali sono i
fattori da ricercare, che possono inficiare la prognosi del bambino con PHS e
coinvolgimento renale. L'importante è sempre una diagnosi precoce e il
ricercare attentamente determinati parametri onde attuare una rapida ed
efficace strategia terapeutica.
Infine c'è il coinvolgimento renale. Nel momento
in cui insorge una glomerulonefrite, che può essere più o meno grave,
automaticamente si passa alla forma complicata, in quanto il rischio di morte è
maggiore così come anche delle recidive. È presente nel 30-40% dei pazienti,
ovvero 1/3 o più dei bambini sviluppa il coinvolgimento renale. Anche qui la
causa è l'infiammazione dei vasi glomerulari. L'insorgenza è acuta e la durata
variabile, anche in funzione della gravità. Nella maggioranza dei casi la
glomerulonefrite è lieve con proteinuria, ematuria microscopica e cilindri
eritrocitari. In questo caso la risoluzione è spontanea. In alcuni casi la
glomerulonefrite è importante, con proteinuria notevole, ematuria macroscopica
ed ipertensione. Questa forma è causa di una insufficienza renale acuta. Nella maggioranza
dei casi la glomerulonefrite compare nei primi mesi dalla presentazione della
PHS. In alcuni casi l'interessamento renale è tardivo e si configura come un
fattore prognostico negativo in quanto predispone ad una malattia renale
cronica. A questo punto ci si potrebbe chiedere se sia possibile prevedere
quale soggetto possa sviluppare una malattia renale cronica e stabilirne anche
la gravità. Sicuramente la proteinuria e l'ematuria sono parametri da ricercare
attraverso l'esame delle urine. Così come anche bisogna valutare bene la
funzione renale, attraverso l'eGFR. Questa tabella riassume quali sono i
fattori da ricercare, che possono inficiare la prognosi del bambino con PHS e
coinvolgimento renale. L'importante è sempre una diagnosi precoce e il
ricercare attentamente determinati parametri onde attuare una rapida ed
efficace strategia terapeutica.

Figura : strategia per la prevenzione della nefrite della PHS.
![]()
 Questo a lato invece è una tabella nella quale
viene riportato uno score, in questo modo è possibile effettuare una stratificazione
del rischio per il singolo paziente. Da notare che nei parametri presi in
considerazione non figura l'interessamento cutaneo, poiché siccome è sempre
presente non risulta essere di ausilio nel determinare la prognosi. Come si può
vedere, per ogni parametro viene dato un punteggio da 0 a 3 e dalla somma dei
singoli punteggi si ottiene uno score, col quale si può correlare la situazione
clinica con la prognosi.
Questo a lato invece è una tabella nella quale
viene riportato uno score, in questo modo è possibile effettuare una stratificazione
del rischio per il singolo paziente. Da notare che nei parametri presi in
considerazione non figura l'interessamento cutaneo, poiché siccome è sempre
presente non risulta essere di ausilio nel determinare la prognosi. Come si può
vedere, per ogni parametro viene dato un punteggio da 0 a 3 e dalla somma dei
singoli punteggi si ottiene uno score, col quale si può correlare la situazione
clinica con la prognosi.
Passiamo adesso agli esami di laboratorio. Siamo in presenza di una vasculite con infiammazione dei piccoli vasi, quindi gli esami ematochimici mostreranno un aumento della VES e della PCR, nonché un aumento della conta leucocitaria, soprattutto neutrofili e basofili. La conta piastrinica risulta essere nella norma. Per la funzione renale, si valutano azotemia e creatininemia. L'imaging è particolarmente utile per la diagnosi della perforazione intestinale e l'invaginazione. La perforazione intestinale si valuterà tramite un esame radiologico mentre per l'invaginazione è fondamentale l'ECO-addome. Per quanto riguarda la ricerca di sangue occulto nelle feci ci fornisce delle indicazioni in merito all'ischemia intestinale. Infine, molto importanti sono le analisi delle urine.
Questo
schema mostra come possa presentarsi l'analisi delle urine al momento della
presentazione della patologia. Come si può notare, ematuria e proteinuria
possono manifestarsi indipendentemente l'una dall'altra. In circa 1/3 dei casi
è presente microematuria e proteinuria al di sotto del range nefrotico, mentre
in 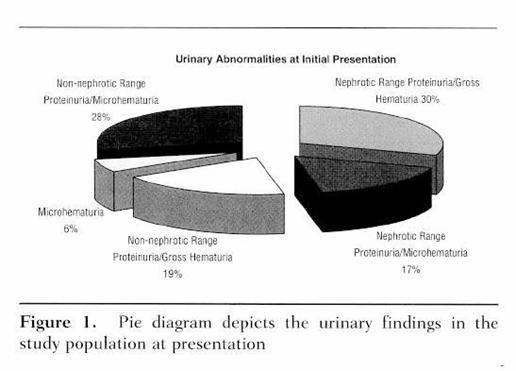 circa il 50% dei casi è presente una proteinuria
nefrosica, accompagnata o meno da macroematuria. Il tutto per dire che, il
quadro urinario può assumere forme miste e non necessariamente presentarsi in
maniera palese con macroematuria e proteinuria massiva.
circa il 50% dei casi è presente una proteinuria
nefrosica, accompagnata o meno da macroematuria. Il tutto per dire che, il
quadro urinario può assumere forme miste e non necessariamente presentarsi in
maniera palese con macroematuria e proteinuria massiva.
![]() Quelli
mostrati sono i quadro anatomopatologici ed immunoistochimici tipici della PHS.
Innanzitutto si può notare l'infiltrato infiammatorio analizzato in differenti
sedi, in quanto stiamo parlando di una patologia sistemica. La causa
dell'infiammazione sono i depositi di IgA, che elicitano una risposta infiammatoria
con il richiamo di neutrofili.
Quelli
mostrati sono i quadro anatomopatologici ed immunoistochimici tipici della PHS.
Innanzitutto si può notare l'infiltrato infiammatorio analizzato in differenti
sedi, in quanto stiamo parlando di una patologia sistemica. La causa
dell'infiammazione sono i depositi di IgA, che elicitano una risposta infiammatoria
con il richiamo di neutrofili.
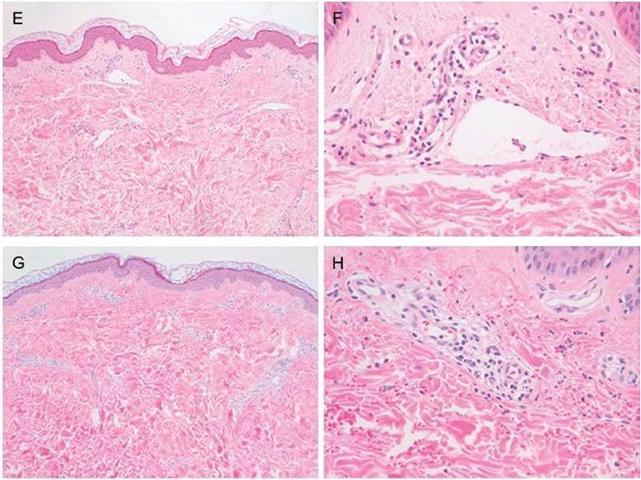
Figura : Quadri anatomo-patologici.
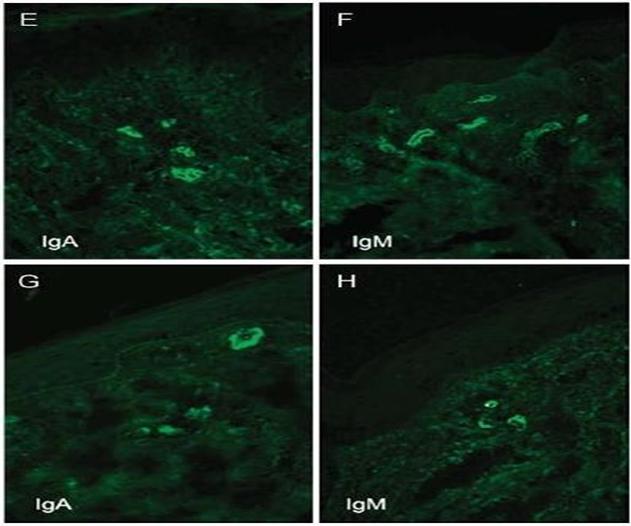
Figura : Quadri immunoistochimici della PHS.
![]()
 Questa tabella è uno score delle manifestazioni
anatomopatologiche che, in base al punteggio, ben si correla con la gravità del
quadro clinico. In poche parole rappresenta la valutazione di una situazione
clinica mediante l'utilizzo di parametri anatomopatologi.
Questa tabella è uno score delle manifestazioni
anatomopatologiche che, in base al punteggio, ben si correla con la gravità del
quadro clinico. In poche parole rappresenta la valutazione di una situazione
clinica mediante l'utilizzo di parametri anatomopatologi.
La diagnosi di PHS si basa sulla presenza di due di questi quattro criteri:
porpora palpabile: lesioni cutanee emorragiche, rilevate, palpabili, in assenza di trombocitopenia;
angina intestinale: dolore addominale diffuso o diagnosi di ischemia intestinale;
biopsia diagnostica: cambiamenti istologici che mostrano granulociti nelle pareti di arteriole o venule;
età pediatrica: età inferiore ai 20 anni all'esordio dei sintomi.
Per quanto riguarda la biopsia, si potrebbe effettuare anche una biopsia renale, tuttavia si ricordi che l'interessamento renale non è sempre presente ed inoltre esistono aree di tessuto più facilmente reperibili, come la cute, quindi la biopsia deve essere effettuata, a maggior ragione che si tratta di soggetti pediatrici, sulla cute. La diagnosi differenziale deve essere posta nei confronti delle altre vasculiti sistemiche e patologie associate con porpora trombocitopenica (porpora trombocitopenica idiopatica, leucemia e LES). Si sottolinei come, un parametro molto importante da valutare al momento della presentazione clinica sia la pressione arteriosa. In caso di ipertensione la prognosi è meno favorevole.
Il bambino deve continuare a fare una dieta regolare, deve essere idratato e bisogna evitare la somministrazione di aspirina, in quanto responsabile della sindrome di Reye. Infine, non bisogna mai sottovalutare il rischio di una recidiva. Esistono fattori che possono predire il rischio di una eventuale ricaduta. Il primo elemento è un'età superiore ai 30 anni, poi seguono una porpora che persiste per più di un mese, dolori addominali, ematuria, una grave leucocitoclasia, la presenza di IgA in assenza di IgM.
Per quanto riguarda la terapia, l'ospedalizzazione è fondamentale, in quanto il bambino può andare incontro ad insufficienza renale, perforazioni intestinali, può andare incontro a tutta una serie di eventi che mettono a repentaglio la sua vita. La terapia di supporto prevede idratazione (modica e non eccessiva). Per le lesioni cutanee la terapia è la somministrazione di glucocorticoidi, prednisone 1 mg per Kg di peso corporeo al giorno, per 1-2 settimane, dopo di che la dose va progressivamente ridotta. Per i dolori e la febbre si somministra il paracetamolo. Una volta attuata la strategia terapeutica, bisogna capire se questa sta avendo successo, andando a valutare gli indici infiammatori, che lentamente si normalizzano, la conta leucocitaria diminuisce, ad eccezione dei neutrofili. Questa anomalia è dovuta alla somministrazione dei glucocorticoidi, che diminuendo la quota di neutrofili che migra nei tessuti determina un'espansione del pool ematico, ma nel lungo termine, la terapia determina una riduzione anche dei neutrofili circolanti. Per il trattamento della situazione renale, ovviamente dipende dal quadro di gravità, prevedendo nei casi più gravi anche la somministrazione di immunosoppressori, antiaggreganti ed anticoagulanti. Nei casi estremi si può consigliare anche il trapianto renale, ma siccome di tratta di una patologia sistemica, ciò non esclude che anche nel rene trapiantato si verifichi lo stesso quadro.
Sindrome di Kawasaki o sindrome muco-cutanea dei linfonodi.
![]()
 La SK è
una vasculite acuta sistemica che colpisce i vasi di medio calibro di tutti i
distretti dell'organismo, autolimitante, ad eziologia sconosciuta,
probabilmente multifattoriale, che colpisce prevalentemente lattanti e bambini.
Al contrario della porpora di SH, che interessa i vasi di piccolo calibro, la
SK interessa invece vasi di medio calibro, soprattutto sul versante arterioso.
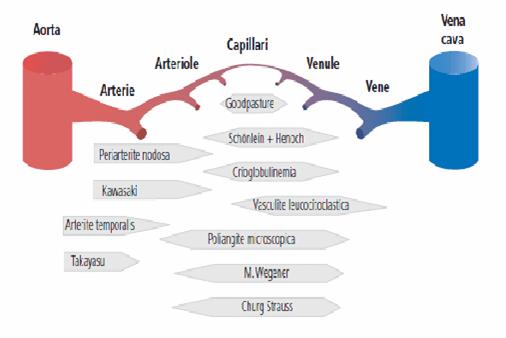
Lo schema mostra le principali vasculiti, riportando anche le sedi che sono
maggiormente colpite. La PAN, la SK, l'arterite temporale e la Takayasu tendono
a colpire i vasi sul versante arterioso di medio calibro.
La SK è
una vasculite acuta sistemica che colpisce i vasi di medio calibro di tutti i
distretti dell'organismo, autolimitante, ad eziologia sconosciuta,
probabilmente multifattoriale, che colpisce prevalentemente lattanti e bambini.
Al contrario della porpora di SH, che interessa i vasi di piccolo calibro, la
SK interessa invece vasi di medio calibro, soprattutto sul versante arterioso.
Lo schema mostra le principali vasculiti, riportando anche le sedi che sono
maggiormente colpite. La PAN, la SK, l'arterite temporale e la Takayasu tendono
a colpire i vasi sul versante arterioso di medio calibro.
Per quanti riguarda l'epidemiologia, i maschi risultano essere maggiormente i più colpiti, con un rapporto M:F di 1.5-1.7. La fascia di età più colpita è rappresentata dai bambini d'età inferiore ai 4 anni, con un picco al secondo anno. Tuttavia, la diagnosi tardiva determina la presenza di soggetti con un'età superiore alla media, ed è facile pensare come un ritardo diagnostico determini anche un maggior rischio di sviluppare complicanze. È una patologia che presenta un andamento endemico, con riaccensioni ogni 2-3 anni e con picco in inverno e in primavera. L'incidenza annuale è di 3-100/100.000 abitanti, con maggiore prevalenza nei bambini di origine asiatica (Giappone). Questi dati riportati derivano dai registri epidemiologici americano e giapponese, mentre in Europa e in Italia la reale incidenza della SK non è perfettamente stimata, data la mancanza di opportuni registri epidemiologici. È la seconda vasculite più frequente nel bambino dopo la porpora di SH. Nonostante manchino dati attendibili della prevalenza in Italia ed in Europa, rappresenta un insidia in ambito diagnostico, e proprio per tale motivo non deve essere sottovaluta, bisogna sapere e conoscere cosa cercare.
Per quanto riguarda l'eziopatogenesi, si suppone che sia una patologia di origine tossinfettiva e immunomediata, che coinvolge superantigeni streptococcici e stafilococcici, con successiva attivazione linfocitaria. Molto spesso la SK esordisce dopo patologie virali. Ma in realtà ancora oggi la reale eziopatogenesi ancora non è conosciuta, ma fattori ambientali e genetici concorrono in sinergia per la determinazione della patologia. Agenti infettivi e superantigeni, raggiungendo ospiti particolarmente suscettibili per motivi genetici, che presentano quindi un'alterazione delle normali risposte immunologiche, determinano una superattivazione immunitaria, con aumento del CD40L, citochine che a loro volta sarebbero i diretti responsabili del danno endoteliale, con innesco di un fenomeno vasculitico coinvolgente anche le coronarie, con la formazione di veri e propri aneurismi.
Da un punto di vista clinico, distinguiamo tre fasi principal: una fase acuta, subacuta e convalescenziale. La fase acuta è caratterizzata da febbre elevata, aspetto sofferente, settico, anoressia ed irritabilità. La febbre può essere molto elevata, con andamento remittente, non responsiva ai normali trattamenti antipiretici. Dopo 8-10 giorni dall'inizio delle febbre compare un rash polimorfo maculo-papuloso, morbilliforme o scarlattiniforme, pruriginoso ed esteso a tutto il corpo. A livello delle congiuntive compare una iperemia congiuntivale non essudativa, che va in diagnosi differenziale con le classiche congiuntiviti dell'età pediatrica. Inoltre vi sono anche lesioni a livello della mucosa orale (lesioni crostose, lingua a fragola, eritema) e cheiliti angolari. I linfonodi cervicali si mostrano palpabili o dolenti. Può essere presente un edema duro al dorso delle mani e dei piedi, tumefazioni fusiformi delle dita, eritema palmare e plantare rosso porpora.
Nella fase subacuta si verificano una desquamazione a larghe lamelle e piastrinosi. Nella fase di convalescenza invece si ha la risoluzione della malattia, con scomparsa dei segni clinici, normalizzazione della VES e presenza di solchi ungueali trasversi caratteristici, dette linee di Beau.
Quindi, ricapitolando, nella fase acuta della SK prevale il picco febbrile, comincia anche il coinvolgimento cardiovascolare, vi è un coinvolgimento della cute, in modo particolare l'eritema palmare e coinvolgimento della congiuntiva e della cavità orale. Nella fase subacuta continua ad esserci la febbre, associata a desquamazione e alle linee ungueali distrofiche. Mentre nella fase di convalescenza persistono le distrofie ungueali ma la febbre regredisce.
La SK coinvolge anche il cuore. Se la patologia viene diagnostica in fase precoce sarà possibile prevenire le eventuali complicanze, soprattutto a livello cardiaco. Siccome stiamo parlando di vasculite, è impossibile non pensare che vi sia un coinvolgimento anche a livello dei vasa vasorum delle coronarie, determinando una miocardite, con predilizione per il tessuto di conduzione. La pancardite si innesca a partire dall'11-25° giorno dall'inzio della patologia, con degenerazione dell'intima delle coronarie e rottura della membrana limitante esterna. Tale quadro evolve verso la formazione di aneurismi coronarici. Nell'ambito delle alterazione delle strutture vascolari del cuore distinguiamo 4 stadi. Nel primo stadio, comprendente i primi 10 giorni di malattia, si verifica un processo vasculitico, perivasculitico dei microvasi e delle piccole arterie, tra cui anche i vasa vasorum delle coronarie. Ciò innesca un quadro di pericardite acuta, miocardite interstiziale, endocardite ed infiammazione del tessuto di conduzione. Nello stadio 2, compreso tra l'11-25° giorno, si verifica una peri e pan-vasculite dei vasi di medio calibro, ed in particolare delle coronarie, con interessamento elettivo dell'intima. In questa fase la rottura della limitante interna può favorire la comparsa di dilatazioni e aneurismi coronarici, soprattutto alle biforcazioni. Tipiche di questa fase sono le lesioni infiammatorie del setto interatriale e interventricolare. Nello stadio 3, compreso tra il 26-30° giorno si assiste alla formazione di trombi, con ispessimento dell'intima delle piccole arterie, anche in assenza di aneurismi preesistenti. Infine, nello stadio 4, dal secondo mese di malattia, si verifica la proliferazione fibroblastica dell'intima, che riempie la zona periferica del sacco aneurismatico, rispettando il lume del vaso con successiva cicatrizzazione e calcificazione.
Per poter fare diagnosi, si utilizza la positività a determinati criteri clinici, che sono:
febbre da più di 5 giorni;
iperemia congiuntivale bilaterale;
alterazioni delle labbra e della cavità orale;
esantema polimorfo;
alterazioni delle estremità;
desquamazione delle dita (entro 2-3 settimane dall'esordio della febbre);
linfoadenopatia cervicale.
Gli esami di laboratorio metteranno in evidenza leucocitosi neutrofila, aumento di VES e PCR, aumento delle AST e ALT, aumento del fibrinogeno. All'ECG si potranno avere delle alterazioni e piastrinosi dopo 10 giorni. Ovviamente, da tenere ben presente, che tutti questi reperti sono completamente aspecifici.
![]()
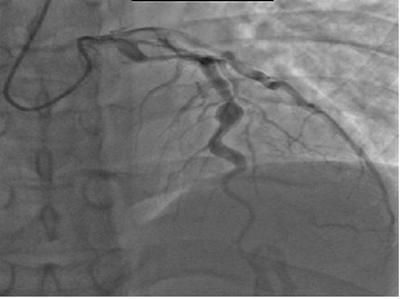 Quindi,
non essendo disponibili test di laboratorio specifici, la SK viene
diagnosticata in base ai criteri clinici e, se presenti, all'osservazione degli
aneurismi coronarici mediante ecocardiografia, angiografia coronarica o TC
delle coronarie. Nel momento in cui si sopsetti una SK, le eventuali
complicanze cardiache devono essere attentamente indagate, ma è ovvio che
l'esecuzione di un esame diagnostico con l'utilizzo di un mdc in un bambino di
4 anni non sia opportuno. Ed è per questo che ci si avvale di metodiche
quali l'ecocardiografia. Nell'immagine
viene mostrata una coronarografia con aneurisma del ramo discendente anteriore
dell'arteria coronaria sinistra. In questi casi, bisogna porre il sospetto che
l'aneurisma sia un esito di una SK misconosciuta. Per quanto riguarda la TC
questa richiede una riduzione della frequenza cardiaca tramite l'utilizzo di un
beta-bloccante e ovviamente tale atteggiamento è improponibile per un bambino
con un'età massima di 4 anni. Per fortuna oggi è possibile effettuare una
diagnosi precoce e applicare di conseguenza il corretto schema terapeutico, per
cui le complicanze a livello cardiaco risultano essere molto rare.
Quindi,
non essendo disponibili test di laboratorio specifici, la SK viene
diagnosticata in base ai criteri clinici e, se presenti, all'osservazione degli
aneurismi coronarici mediante ecocardiografia, angiografia coronarica o TC
delle coronarie. Nel momento in cui si sopsetti una SK, le eventuali
complicanze cardiache devono essere attentamente indagate, ma è ovvio che
l'esecuzione di un esame diagnostico con l'utilizzo di un mdc in un bambino di
4 anni non sia opportuno. Ed è per questo che ci si avvale di metodiche
quali l'ecocardiografia. Nell'immagine
viene mostrata una coronarografia con aneurisma del ramo discendente anteriore
dell'arteria coronaria sinistra. In questi casi, bisogna porre il sospetto che
l'aneurisma sia un esito di una SK misconosciuta. Per quanto riguarda la TC
questa richiede una riduzione della frequenza cardiaca tramite l'utilizzo di un
beta-bloccante e ovviamente tale atteggiamento è improponibile per un bambino
con un'età massima di 4 anni. Per fortuna oggi è possibile effettuare una
diagnosi precoce e applicare di conseguenza il corretto schema terapeutico, per
cui le complicanze a livello cardiaco risultano essere molto rare.
Poiché si tratta di una patologia sistemica, gli aneurismi possono coinvolgere anche altri distretti, quali arterie succlavie, brachiali, ascellari, iliache, renali, mesenteriche o intercostali. Pertanto è considerata obbligatoria l'esplorazione anche di tutti questi distretti con appropriate ecografie.
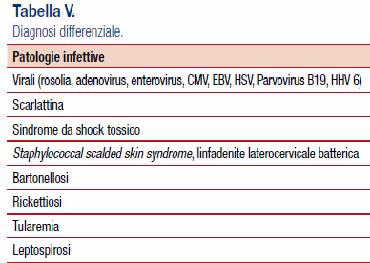
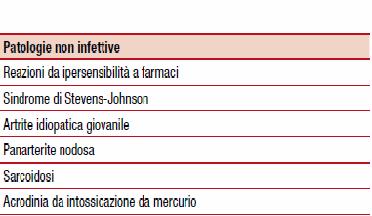
Per quanto riguarda la diagnosi differenziale, questa deve essere posta sia nei confronti patologie infettive che non. L'interessamento cutaneo con l'esantema deve essere posto in diagnosi differenziale con tutte le malattie esantematiche tipiche dell'infanzia, quali la scarlattina, infezioni virali, ecc. Tra le patologie non infettive citiamo la sarcoidosi, per via dell'interessamento della componente elettrica del cuore, reazioni allergiche, ecc
 Per
quanto riguarda la prognosi, questa è una patologia che tende a risolversi in
maniera spontanea. Tuttavia, il 5-10% dei pazienti sviluppa complicanze gravi,
per lo più cardiologiche, e l'1% è afflitto da complicanze letali. Nella prima
fase si possono osservare miocardite e pericardite. Successivamente, possono
comparire infarto del miocardio e rottura di aneurismi delle coronarie. Altre
complicanze sono l'uretrite, artropatie, meningite asettica e ittero ostruttivo.
Per
quanto riguarda la prognosi, questa è una patologia che tende a risolversi in
maniera spontanea. Tuttavia, il 5-10% dei pazienti sviluppa complicanze gravi,
per lo più cardiologiche, e l'1% è afflitto da complicanze letali. Nella prima
fase si possono osservare miocardite e pericardite. Successivamente, possono
comparire infarto del miocardio e rottura di aneurismi delle coronarie. Altre
complicanze sono l'uretrite, artropatie, meningite asettica e ittero ostruttivo.
La terapia si basa sulla somministrazione di:
 immunoglobuline
per via ev;
immunoglobuline
per via ev;
aspirina;
infliximab.
![]() Le
immunoglobuline rappresentano un valido ausilio terapeutico, esercitando
un'azione antinfiammatoria e attivazione dei linfociti T-suppressor, riuscendo
a prevenire le complicazne aneurismatiche della SK. L'aspirina può essere
utilizzata sia nelle fasi acute che avanzate. Nelle fasi acute vengono utilizzati
elevati dosaggi per sfruttare l'effetto antinfiammatorio, mentre nelle fasi
avanzate l'aspirina viene somministrata a basse dosi per innescare il suo
effetto antiaggregante. Nella somministrazione dell'aspirina nel bambino,
particolare attenzione deve essere posta per il possibile sviluppo di una
sindrome di Reye. Questi due presidi, immunoglobuline e aspirina, sono in grado
di prevenire tutte le complicanze, soprattutto quelle a livello cardiaco. In
caso di soggetti non responders, è possibile utilizzare l'infliximab, un
anti-TNF-alpha. L'infliximab in questi casi rappresenta una valida alternativa.
Le
immunoglobuline rappresentano un valido ausilio terapeutico, esercitando
un'azione antinfiammatoria e attivazione dei linfociti T-suppressor, riuscendo
a prevenire le complicazne aneurismatiche della SK. L'aspirina può essere
utilizzata sia nelle fasi acute che avanzate. Nelle fasi acute vengono utilizzati
elevati dosaggi per sfruttare l'effetto antinfiammatorio, mentre nelle fasi
avanzate l'aspirina viene somministrata a basse dosi per innescare il suo
effetto antiaggregante. Nella somministrazione dell'aspirina nel bambino,
particolare attenzione deve essere posta per il possibile sviluppo di una
sindrome di Reye. Questi due presidi, immunoglobuline e aspirina, sono in grado
di prevenire tutte le complicanze, soprattutto quelle a livello cardiaco. In
caso di soggetti non responders, è possibile utilizzare l'infliximab, un
anti-TNF-alpha. L'infliximab in questi casi rappresenta una valida alternativa.
|
| Appunti su: porpora schonlein, esantema morbilliforme, porpora di schonlein, borrelia e porpora, porpora malattia, |
|
| Appunti Bellezza |  |
| Tesine Nutrizione |  |
| Lezioni Bambini |  |