|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 2169 | Gradito: |
Leggi anche appunti:FavismoFAVISMO Che cos'è il favismo Il favismo è un difetto congenito di Scialoadeniti autoimmuni - Sindrome di SjögrenScialoadeniti autoimmuni - Sindrome di Sjögren È una patologia sistemica, Cenni di elettroencefalografiaCenni di elettroencefalografia L'elettroencefalografia (EEG) è una tecnica per la registrazione non invasiva |
 |
 |
ONCOLOGIA PEDIATRICA
Un bambino ogni 600 sviluppa un tumore nei primi 15 anni di vita: ogni anno in Italia vengono diagnosticati circa 1400 casi di tumori; un pediatra diagnostica nella sua vita professionale 1-5 casi di tumori maligni o di leucemie.
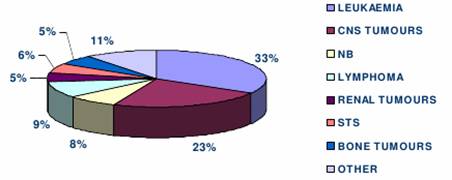 La frequenza dei vari tipi di tumore è:
La frequenza dei vari tipi di tumore è:
leucemie 33%;
tumori del SNC 23%;
linfomi 9%;
neuroblastoma 8%;
sarcomi dei tessuti molli 6%;
neuroblastoma 5%;
![]() tumori
dell'osso 5%;
tumori
dell'osso 5%;
altro 11%.
Eziologia: i fattori di rischio per lo sviluppo tumorale sono l'esposizione (anche in utero) a sostanze chimiche, radiazioni ionizzanti, inquinamento ambientale e farmaci antiblastici; anche i virus (HBV, HCV, HIV e EBV) aumentano il rischio. In età pediatrica tuttavia i tumori possono più di frequente che nell'adulto essere spia di alterazioni ereditarie: in particolare se sono bilaterali, vengono diagnosticati in età più precoce rispetto alla media e se c'è storia famigliare positiva. La quota di tumori ereditari è limitata (3-5%), tuttavia, i portatori di geni responsabili dello sviluppo di neoplasie ereditarie hanno una probabilità di ammalare molto elevata (RB1: RR=1000).
La frazione di casi ereditari mostra grande variabilità tra i diversi tipi di tumori. Le sindromi cromosomiche possono aumentare il rischio di tumori: trisomia 21 leucemia/linfoma RR 20, sd di Morris e Klinefelter aumento del rischio tumori delle cellule germinali. Vi sono anche sindromi in cui lo sviluppo di un tipo di tumore è parte integrante del quadro clinico:
Sindrome WAGR: Aniridia, Anomalie Genitali, Ritardo Mentale e Tumore di Wilms: delezione 11p13, gene WT1. I bambini con aniridia sporadica devono essere sottoposti ad analisi cromosomica;
Sindrome di Denys-Drash: nefropatia congenita, tumore di Wilms e stati intersessuali. AD, missense codon WT1; proposta di screening con eco addome ogni 4 mesi fino all'età di 5 anni;
Sindrome di Beckwith-Wiedermann: disomia uni parentale paterna 15q11; il 7% dei bambini affetti sviluppa un tumore nel primi 5 anni (tumore di Wilms, epatoblastoma, neuroblastoma).
Le mutazioni a carico dei geni preposti al controllo della proliferazione (oncogeni od oncosoppressori) configurano quadri caratteristici in cui lo sviluppo di uno o più tumori è pressoché certo:
Retinoblastoma: mutazione del gene RB1 localizzato nella regione 13q14, interviene nel 40% dei retinoblastomi come mutazione germline (i restanti sono dovuti a mutazioni de novo). Il rischio di ricorrenza nei figli di genitori affetti da RB è del 45%, e c'è maggior rischio di tumori maligni secondari. È possibile la diagnosi prenatale, in più si monitorano i bambini a rischio con fundus ogni 2-3 mesi per 2 anni, poi ogni 4-6 mesi;
Sindrome di Li-Fraumeni: nel 65-80% mutazione germline di p53; gli individui affetti presentano sarcomi, tumori mammari premenopausa, leucemie e tumori cerebrali entro i 45 anni di vita. I portatori del gene sviluppano più tumori sincroni o metacroni non associati alle terapie (RR: 5.3). La mutazione è stata rilevata in oltre il 50% dei bambini con carcinoma adrenocorticale e nel 10% con osteosarcoma e rabdomiosarcoma;
Poliposi famigliare del colon: mutazione gene APC in 5p21.22; colpisce 1:8.000. I bambini delle famiglie FAP sono ad alto rischio di sviluppare un HB: 1 su 250 (RR 800); l'individuazione precoce dei bambini portatori è importante più che altro per impostare lo screening con rettosigmoidoscopia a partire da 8-10 anni;
Neurofibromatosi tipo 1: mutazione gene NF1 in 17q11.2. La frequenza nella popolazione generale è di 1:2500, con elevata percentuale di mutazioni de novo. C'è un eccesso di tumori benigni (neurofibromi), gliomi del chiasma (15% dei bambini con NF1), tumori maligni delle guaine nervose, sarcomi, LNLA (LNLA:LLA 20:9 vs 1:4), feocromocitomi;
Neurofibromatosi tipo 2: mutazione gene NF2 sul cromosoma 22; predispone a meningiomi e gliomi del nervo acustico;
Malattia di von Hippel-Lindau (VHL): mutazione gene 3p25-26, mutazione germline 100%. È associata a emangioblastomi cerebellari, angiomi retinici, carcinomi renali e pancreatici e feocromocitomi. Screening con eco addome, fundus e catecolamine urinarie ogni anno;
Ataxia-teleangectasia: gene ATM su 11q22-q23; eccesso di leucemie/linfomi e tumori (mammari) negli eterozigoti.
Tumori del SNC
25-30% dei tumori in età pediatrica; sono i tumori solidi più frequenti e hanno picco tra 5 e 10 anni.
Clinica: secondaria all'ipertensione endocranica; cefalea, vomito a getto, letargia. Possono esservi segni di interessamento focale quali diplopia, atassia, ritardo di crescita e alterazioni endocrine nei tumori sellari. Sotto i 2 anni si osserva irrequietezza, tensione della fontanella, occhi "a sole calante".
Localizzazione: sopratentoriali nel 40% (astrocitoma, ependimoma, glioblastoma, meningioma, adenoma pituitario, gliomi) e sottotentoriali nel 60% (medulloblastoma 25%, astrocitoma 18%, meningioma, astrocitoma, ependimoma, glioblastoma).
Diagnosi: esame neurologico, oftalmoscopico, oculistico, TC con mdc, RMN con mdc, angiografia, EEG, esame del LCR, biopsia.
Terapia: la chirurgia è fondamentale, associata o meno a radio- e chemioterapia.
Tumore di Wilms
Tumore embrionario maligno del rene, costituito da combinazioni variabili di elementi di blastomatosi, stromali ed epiteliali. Di solito si manifesta in bambini d'età < 5 anni, ma a volte anche in bambini più grandi e raramente in età adulta. In circa il 4% dei casi il tumore di Wilms compare contemporaneamente in entrambi i reni.
Clinica: massa addominale palpabile, dolore addominale, ematuria, febbre, anoressia, nausea e vomito. L'ematuria, che compare nel 15-20% dei casi, indica l'interessamento del sistema collettore. L'ipertensione può essere secondaria a ischemia da compressione del peduncolo o del parenchima.
Diagnosi: ecografia addominale, TC addome, Rx torace e TC per escludere metastasi.
Terapia: è indicata l'esplorazione chirurgica immediata della lesione, potenzialmente asportabile, e del rene controlaterale. La chemioterapia con actinomicina. D e vincristina, con o senza radioterapia, è subordinata allo stadio del tumore. I bambini con stadio avanzato sono trattati anche con doxorubicina.
Prognosi: dipende dalle caratteristiche istologiche del tumore, dallo stadio di quest'ultimo al momento della diagnosi e dall'età del paziente (la prognosi è migliore se il paziente è più giovane).
Neuroblastoma
Tumore maligno solido che origina di solito dalle ghiandole surrenali ma anche da qualsiasi altra parte della catena simpatica extra-surrenalica, compresa quella in sede retroperitoneale o toracica. 75% <5 anni. Circa il 65% dei neuroblastomi è a partenza addominale mentre il 15-20% compare a livello toracico; il rimanente 15% interessa altri distretti come il collo e la pelvi. Molto raramente il neuroblastoma si può manifestare come tumore primitivo del SNC. La maggior parte dei neuroblastomi produce catecolamine, rilevabili per la presenza di livelli elevati di metaboliti urinari.
Clinica: dipende dalla sede; si possono riscontrare una massa addominale palpabile o dei problemi respiratori dovuti a un interessamento toracico. Segni e sintomi d'esordio possono essere causati da metastasi: epatomegalia, dolore osseo, anemia, petecchie e leucopenia. I bambini possono manifestare delle sindromi paraneoplastiche come opsoclono-mioclono, diarrea secretoria o ipertensione, nonché deficit neurologici focali dovuti all'estensione diretta del tumore nel canale midollare.
Diagnosi: ecografia e TC. Per la ricerca di metastasi si ricorre ad aspirato midollare da sedi differenti, radiografia dell'intero scheletro, scintigrafia ossea, TC e talvolta scintigrafia con I-metaiodobenzilguanidina. La concentrazione urinaria di acido vanilmandelico (VMA) è elevata nel 65% dei pazienti; la scintigrafia e un livello alto di acido omovanillico permettono di individuare più del 90% dei pazienti. Quando il tumore viene asportato, una parte di esso deve essere sottoposta ad analisi dell'indice mitotico e dell'amplificazione dell'oncogene MYCN.
Terapia: rimozione chirurgica, chemioterapia sopra all'anno di vita con vincristina, ciclofosfamide, doxorubicina, cisplatino, carboplatino ed etoposide; la radioterapia è necessaria per neoplasie avanzate.
Prognosi: la possibilità della rimozione chirurgica della massa tumorale primitiva migliora molto la prognosi. Sono fattori di prognosi positiva l'età al di sotto di 1 anno, uno stadio basso e l'assenza di amplificazione dell'oncogene MYCN.
Retinoblastoma
Tumore maligno che prende origine dalla retina immatura. 1:15.000-30.000 nati vivi e rappresenta circa il 2% dei tumori maligni dell'infanzia.
Diagnosi: di solito si effettua dal terzo al quarto anno di vita quando compare un riflesso bianco pupillare (leucocoria o pupilla a occhio di gatto) o strabismo. Devono essere esaminati attentamente entrambi i fondi oculari con l'oftalmoscopia indiretta a pupille ben dilatate e in anestesia generale. I tumori appaiono nella retina come rilevature bianco-grigiastre singole o multiple; parti tumorali possono essere visibili nel corpo vitreo. In quasi tutti i tumori si possono evidenziare calcificazioni mediante la TC.
Screening: i componenti più prossimi della famiglia di ogni bambino affetto devono essere sottoposti almeno una volta a esame oftalmologico per escludere il retinoblastoma (nei bambini piccoli) o il retinocitoma (nelle persone più grandi). Le sonde di DNA ricombinante possono fornire un aiuto per individuare i portatori asintomatici.
Terapia: fino a qualche anno fa il retinoblastoma veniva soltanto enucleato dalla sede. L'aumento dei casi affetti bilateralmente, ha dovuto far pensare alla possibilità di una terapia conservativa dell'occhio pur aggredendo il tumore fino a farlo regredire totalmente. Si è quindi passati ad una mentalità di terapia conservativa del bulbo oculare, nel rispetto della vita del paziente.
Attualmente i trattamenti combinati locali (fotocoagulazione mediante laser, criocoagulazione, chirurgia e radioterapia) e sistemici (chemioterapia), tra loro associati, permettono non solo un'elevata percentuale di guarigione, ma anche la preservazione dell'organo della vista.
Il trattamento è composto dall'associazione di cicli di chemioterapia ed aggressione del tumore con laser o crioterapia. La scelta del tipo di trattamento è giustificata dal volume e dalla localizzazione della massa tumorale. Contemporaneamente si deve esaminare il LCR e il midollo osseo per la ricerca di eventuali cellule maligne. I pazienti con retinoblastoma ereditario presentano un'incidenza aumentata di un secondo tumore maligno, il 50% dei quali ha origine nella zona irradiata. Entro 30 anni dalla diagnosi, il 70% di essi ha sviluppato una seconda neoplasia maligna.
Rabdomiosarcoma
Neoplasia maligna che insorge dal tessuto embrionario che da origine ai muscoli striati. 4-5 nuovi casi all'anno ogni 1.000.000 di bambini di età inferiore a 15 anni, con picco di incidenza 2-5 anni di età. Prevalenza nel sesso maschile con un rapporto maschi/femmine di 1,5:1.
Clinica: i sintomi variano a seconda della sede colpita:
testa e collo (30-65%): disfonia, disfagia, tumefazione, sinusiti ed epistassi recidivanti, esoftalmo;
tronco ed estremità (10-25%): massa palpabile, sintomi da compressione sul midollo spinale;
apparato genito-urinario (20%): disturbi della minzione, ematuria, sanguinamento vaginale.
Diagnosi: va eseguito prelievo bioptico per l'esame istologico.
Terapia: chirurgica solo se si ritiene possibile l'asportazione radicale; al contrario va eseguito prima un ciclo di polichemioterapia, seguito dall'asportazione della massa.
Prognosi: sopravvivenza globale a 2 anni: 65%. Sopravvivenza libera da malattia a 2 anni: 55%.
|
| Appunti su: oncologia pediatrica appunti, appunti di oncoematologia pediatrica, incidenza gliomi etC3A0 pediatrica, retinocitoma, |
|
| Appunti Bambini |  |
| Tesine Nutrizione |  |
| Lezioni Bellezza |  |