|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 4013 | Gradito: |
Leggi anche appunti:Dermatite eczematosa cronica - inquadramento clinicoDERMATITE ECZEMATOSA CRONICA - INQUADRAMENTO CLINICO Con Uretriti non gonococcicheUretriti non gonococciche Sono infezioni a trasmissione sessuale caratterizzate Dermatosi vescicoloseDERMATOSI VESCICOLOSE Herpes simplex E' una dermatosi causata da HSV un |
 |
 |
MALATTIE METABOLICHE - BAMBINO
La diagnosi delle malattie metaboliche viene effettuata mediante spettri o mediante cromatografia. Alcuni elementi possono presentare la stessa migrazione elettroforetica o cromatografica e si può generare un errore. Oggi la diagnosi è molto più precisa, viene effettuata mediante spettrometri di massa che bombardano la molecola, si valuta lo spettro della molecola e si identifica in maniera univoca la molecola responsabile.
Le malattie metaboliche vanno sospettate in presenza di letargia, iperammoniemia, crisi ipoglicemiche, acidosi, grave epatopatia o quadro neurologico acuto.
Il sospetto diagnostico può salvare la vita dei pazienti. La diagnosi corretta può consentire di fornire un consiglio genetico alle famiglie.
Sono malattie molto rare soltanto se le consideriamo singolarmente, non sono rare come gruppi. Se si cercano e sono disponibili tecniche diagnostiche adatte si trovano. Si possono portare i campioni presso laboratori centralizzati specializzati, ad esempio negli USA, Inghilterra, Francia.
Hanno distribuzioni etniche diverse, ad esempio alcune malattie metaboliche sono più frequenti tra gli Ebrei Ashkenazi. Il numero delle amminoacidemie è aumentato nel corso degli anni con l'evoluzione delle indagini strumentali. Inizialmente le malattie metaboliche venivano diagnosticate mediante l'analisi chimica-clinica dell'amminoacido, ad esempio si ritrovava la tirosina e veniva diagnosticata la tirosinemia. Successivamente con l'analizzatore automatico di amminoacidi venivano valutati diversi amminoacidi in corsa. Gli amminoacidi vengono colorati con la ninidrina e a seconda dell'RF di migrazione elettroforetica o cromatografica, si valuta in base al tempo di eluizione per quel momento il picco di una determinata sostanza. Quest'esame viene effettuato analizzando un liquido biologico ad esempio sangue, urina LCS ecc.
La GC-MS, gascromatografia con spettrometria di massa, permette di eluire e separare i vari metaboliti mediante una colonna sotto pressione (LC-MS = cromatografia liquida-spettrometria di massa) oppure mediante la gascromatografia utilizzando una colonna cromatografica per la separazione degli AA che viene bombardata con lo spettro di massa e permette l'identificazione delle singole molecole.
Esistono quadri di presentazione generali che possono indirizzare verso gruppi di malattie. Di solito le malattie metaboliche si presentano con quadri di grave encefalopatia, grave epatopatia, insufficienza renale o tubulopatia ecc
La diagnosi è importante perché molte malattie metaboliche sono attualmente curabili (esempio NTBC per la tirosinemia). I più gravi difetti congeniti del metabolismo si presentano solitamente nel periodo neonatale o nei mesi successivi. Il trattamento precoce previene gli effetti deleteri di queste condizioni, in particolare sul SNC. Inoltre una diagnosi è necessaria per il consiglio genetico; si conservano i campioni biologici anche se il bambino è deceduto per arrivare alla diagnosi, questo è importante per la programmazione di una nuova gravidanza e per effettuare una diagnosi pre-natale.
La diagnostica di laboratorio è molto sofisticata, però in molti casi bastano poche indagini di laboratorio di primo livello (EAB, ac. lattico) per identificare il gruppo di malattie.
Definizione
La malattia metabolica è una condizione patologica che si verifica in mancanza di un enzima che determina un aumento a monte del substrato e una riduzione a valle del catabolita.
In genere il substrato è tossico, ad esempio il succinil-acetone nei confronti di diversi apparati. Nella fenilchetonuria aumenta a monte la fenilalanina mentre a valle in seguito al deficit di fenilalanina idrossilasi viene a mancare la tirosina che determina un quadro di anemia, discheratosi cutanea, sofferenza alla luce in quanto la tirosina è presente nella sintesi della melanina.
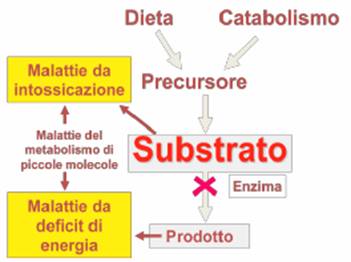 Possono verificarsi delle malattie da
intossicazione per accumulo del substrato a monte e condizioni di carenza per
il prodotto dell'enzima deficitario a valle.
Possono verificarsi delle malattie da
intossicazione per accumulo del substrato a monte e condizioni di carenza per
il prodotto dell'enzima deficitario a valle.
Classificazione
È importante sapersi orientare tra le varie malattie causate da errori del metabolismo.
Possiamo classificare le malattie metaboliche mediante una classificazione biochimica ed una classificazione clinica.
![]() In
generale si possono avere malattie da intossicazione per l'accumulo del
substrato e malattie da deficit di energia per la mancanza di prodotto a valle.
Ad esempio possono essere presenti due situazioni contemporanee: tirosinemia a
monte ed ipoglicemia a valle.
In
generale si possono avere malattie da intossicazione per l'accumulo del
substrato e malattie da deficit di energia per la mancanza di prodotto a valle.
Ad esempio possono essere presenti due situazioni contemporanee: tirosinemia a
monte ed ipoglicemia a valle.
Possiamo suddividere le malattie metaboliche in:
Malattie del metabolismo dei carboidrati e del metabolismo energetico;
Acidemie organiche;
Difetti del ciclo dell'urea;
Malattie del metabolismo degli aminoacidi
L'accumulo del substrato può avvenire all'interno dei lisosomi, dei perossisomi, oppure possono essere presenti altre patologie come i difetti di glicosilazione (CDG), un gruppo di patologie recentemente introdotto caratterizzato da un coinvolgimento multi-organo (SNC, fegato, cute, rene).
Altre patologie più rare sono:
difetti di biosintesi del colesterolo;
difetti del metabolismo di purine e pirimidine;
malattie del metabolismo dei neurotrasmettitori, etc..
La maggior parte degli errori del metabolismo viene trasmessa con modalità AR per cui un'anamnesi di consanguineità e/o di decesso nel periodo neonatale aumenta l'indice di sospetto.
Malattie del metabolismo degli Amminoacidi
Le malattie del metabolismo degli amminoacidi più frequenti sono:
fenilchetonuria;
omocistinuria;
leucinosi;
tirosinemia.
Fenilchetonuria (PKU)
E' importante effettuare lo screening di questa malattia in quanto è frequente e, se diagnosticata, può essere curata in modo da evitare un deficit neurologico permanente. Inoltre lo screening è importante per la programmazione delle gravidanze future per la mamma. Il test di screening viene effettuato mediante un prelievo di sangue dal tallone che viene inviato al centro screening.
E' raccomandabile prelevare il campione ematico nelle prime 24-48h di vita dopo l'assunzione di proteine alimentari per evitare falsi negativi. Si procede con la valutazione della concentrazione di fenilalanina su un dischetto dove viene eluito il sangue. Si usa il Guthrie test che consiste in un test di inibizione della crescita batterica in base alla concentrazione di fenilalanina. La fenilalanina è in grado di inibire la crescita di alcuni batteri. Questo test non richiede particolari macchinari. Possono anche essere usati più semplicemente degli aminoacid analyzer o test di tipo biochimico.
Epidemiologia: presenta una modalità di trasmissione autosomica recessiva, con una frequenza di 1: 10'000.
Eziopatogenesi: Vi è un difetto enzimatico della fenilalanina idrossilasi che metabolizza la fenilalanina in tirosina; il gene della fenilalanina idrossilasi è localizzato sul cromosoma 12q24.1. I cataboliti ac. 2-OH-fenil-acetico, ac. fenil-acetico e acido fenil-lattico sono responsabili dell'odore pungente delle urine.
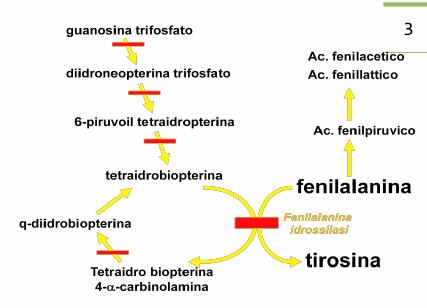 La fenilchetonuria si può verificare anche in
presenza di un'alterazione della tetra-biopterina reduttasi sempre coinvolta
nella reazione di conversione della fenilalanina in tirosina.
La fenilchetonuria si può verificare anche in
presenza di un'alterazione della tetra-biopterina reduttasi sempre coinvolta
nella reazione di conversione della fenilalanina in tirosina.
![]() Quadro clinico: ritardo psicomotorio, compromissione neurologica, convulsioni,
vomito, eczema, cute e capelli chiari (per la mancanza di tirosina), presenza
di problemi visivi. Il quadro biochimico è caratterizzato da un aumento di Phe
plasmatica ed aumentata escrezione urinaria di fenilchetoni, responsabili della
positività al test con il percloruro ferrico (FeCl3). La presenza di
percloruro ferrico nelle urine indica un quadro ormai irreversibile dal punto
di vista neurologico. I fenilchetoni nelle urine non sono presenti in età
neonatale perché il neonato non ha mangiato per un periodo così lungo da avere
un'escrezione urinaria di fenilchetoni.
Quadro clinico: ritardo psicomotorio, compromissione neurologica, convulsioni,
vomito, eczema, cute e capelli chiari (per la mancanza di tirosina), presenza
di problemi visivi. Il quadro biochimico è caratterizzato da un aumento di Phe
plasmatica ed aumentata escrezione urinaria di fenilchetoni, responsabili della
positività al test con il percloruro ferrico (FeCl3). La presenza di
percloruro ferrico nelle urine indica un quadro ormai irreversibile dal punto
di vista neurologico. I fenilchetoni nelle urine non sono presenti in età
neonatale perché il neonato non ha mangiato per un periodo così lungo da avere
un'escrezione urinaria di fenilchetoni.
Dopo aver valutato i livelli di fenilalanina plasmatica può essere effettuata una analisi molecolare. Questa ulteriore indagine è importante per la programmazione delle gravidanza future. Il gene della PAH è stato clonato. Le mutazioni più frequenti sono: R261Q, IVS-546, L48S.
Il paziente può presentare la PKU classica quando i livelli di fenilalanina plasmatica sono <20mg/dL oppure una forma lieve di iper-fenilalaninemia quando i livelli di fenilalanina plasmatica sono circa 2-6mg/dL., questi ultimi presentano un lieve deficit di fenilalanina idrossilasi o della tetra-biopterina (BH4).
Iper-fenilalaninemia da deficit del cofattore BH : Bambini con difetto localizzato su uno degli enzimi necessari per la produzione o riciclaggio del cofattore BH4. Presentano un rapido deterioramento neurologico malgrado l'adeguato controllo della fenilalanina plasmatica in seguito alla diagnosi di PKU.
BH4 è il cofattore anche per la tirosina e la triptofano idrossilasi che svolgono un ruolo essenziale nella biosintesi della dopamina e della serotonina. BH4 è anche cofattore per la ossido nitrico sintetasi.
Questi bambini presentano manifestazioni neurologiche come la perdita di controllo della testa, ipotonia del tronco, presentano anche scialorrea, difficoltà nella deglutizione, convulsioni mio-cloniche. Questi si sviluppano dopo i 3 mesi di età malgrado una dieta adeguata.
Questi pazienti sviluppano iperprolattinemia che può essere dovuta alla carenza di dopamina nell'area ipotalamica.
Terapia
Riduzione dell'apporto dietetico di fenilalanina per mantenere la Phe post-prandiale tra 2 e 6 mg/dl (oltre i 10 anni tra 2 e 8 mg/dl). La dieta a basso contenuto di fenilalanina nel neonato viene effettuata con latte speciale. Successivamente si procede con una dieta a basso contenuto proteico in grado di mantenere i livelli di fenilalanina plasmatica 5-10 mg/dL. L'interruzione della terapia può causare, anche negli adulti, deterioramento del QI e del rendimento cognitivo pertanto si consiglia di seguire la dieta per tutta la vita. La somministrazione orale del cofattore tetra-biopterina (BH4) a pazienti con lieve iper-fenilalaninemia causata da un deficit della fenilalanina idrossilasi può ridurre i livelli plasmatici di fenilalanina;
Alimenti speciali e integratori;
Mantenere un adeguato stato nutrizionale;
Mantenere una normale crescita.
Prevenire il ritardo mentale: è importante far maturare il SNC così da avere una BEE meno permeabile. Anche un paziente eterozigote può presentare un deterioramento neurologico, ad es. un paziente eterozigote con madre affetta da iper-fenilalaninemia (non corretta con la dieta), in quanto il feto, durante la vita intra-uterina, si troverà contemporaneamente ad avere una quota enzimatica inferiore rispetto al soggetto sano e a dover provvedere anche all'iper-fenilalaninemia materna.
Leucinosi
La Leucinosi, o malattia delle urine a sciroppo d'acero rappresenta uno dei più frequenti difetti del metabolismo degli aminoacidi ramificati (leucina, isoleucina e valina).
Epidemiologia: presenta una frequenza di 1:100'000 - 1: 300'000, con una modalità di trasmissione autosomica recessiva.
Eziopatogenesi: Il difetto di base risiede nel complesso enzimatico deidrogenasi degli aminoacidi a catena ramificata. La decarbossilazione della leucina, isoleucina e valina è realizzata da un complesso sistema enzimatico (α-chetoacido deidrogenasi a catena ramificata) che utilizza la tiamina pirofosfato (vitamina B1) come coenzima. Gli amminoacidi maggiormente colpiti sono valina, leucina e isoleucina.
Quadro clinico: Si distinguono diverse forme di leucinosi:
acuta neonatale o forma classica (attività residua inferiore o uguale all'1%);
subacuta o intermittente, con esordio nei primi mesi di vita o dopo il primo anno (attività residua dal 2 al 20%);
tiamino-sensibile (deficit della subunità E1 di cui la tiamina è cofattore enzimatico).
Il quadro clinico della forma neonatale classica è simile ad altre acidosi organiche con comparsa, dopo un intervallo libero da sintomi, di suzione torpida, rifiuto dell'alimentazione, vomito, alterazione del tono muscolare, fontanella pulsante, convulsioni, insufficienza respiratoria e coma. Se non prontamente diagnosticati e trattata, il decesso dei pazienti avviene generalmente in pochi giorni.
Nelle forme subacute e in quelle intermittenti, l'età di esordio è variabile da alcuni mesi ad anni dopo la nascita. In questi casi è caratteristica la completa assenza di sintomi neurologici anche per lunghi periodi, con episodi intermittenti di atassia o crisi di scompenso metabolico.
Lo scompenso è indotto dal catabolismo proteico endogeno, durante stress infettivi e prolungati digiuni, o da un eccessiva introduzione di proteine con l'alimentazione.
La forma vitamino-sensibile ha un decorso clinico sovrapponibile a quella classica, ma risponde rapidamente alla somministrazione di tiamina ad alte dosi con una drammatica ed immediata risoluzione della crisi, sia dal punto di vista clinico che biochimico.
Diagnosi di laboratorio: I reperti di laboratorio sono aspecifici fatta eccezione per l'acidosi metabolica. Importanti sono i test qualitativi urinari (DNPH). L'urina contiene elevate concentrazioni di leucina, isoleucina e valina e dei rispettivi chetoacidi che possono essere individuati aggiungendo all'urina qualche goccia di reagente 2,4-dinitrofenilidrazina (allo 0,1% in 0,1 N di HCl).
L'aminoacidemia quantitativa è caratterizzata dall'aumento di ILE, LEU, VAL. Il prelievo va effettuato 2-4 ore dopo un pasto o seguendo standard locali.
Terapia
Riduzione dell'apporto dietetico di aminoacidi ramificati per mantenere i valori plasmatici di questi aminoacidi nella norma;
Alimenti speciali e integratori;
Mantenere un adeguato stato nutrizionale;
Mantenere una normale crescita, poiché questi amminoacidi non possono essere sintetizzati endogenamente, piccole dosi dovrebbero essere aggiunte alla dieta, titolandole con precisione mediante frequenti analisi dei livelli plasmatici degli amminoacidi;
Prevenire il ritardo mentale, per cui i pazienti dovrebbero attenersi alla dieta per tutta la vita;
Il trattamento dell'episodio acuto ha come obbiettivo l'idratazione e la rapida rimozione degli AA a catena ramificata e dei loro metaboliti dai tessuti e dai liquidi corporei.
La prognosi a lungo tempo resta riservata. Una grave cheto-acidosi ed un edema cerebrale che possono insorgere in condizioni stressanti (infezioni o interventi chirurgici) possono mettere in pericolo la vita del paziente.
Tirosinemia
La tirosina, ottenuta per ingestione di proteine e da sintesi endogena della fenilalanina, è utilizzata per la sintesi proteica ed è precursore della dopamina, della noradrenalina, dell'adrenalina e della melanina.
Epidemiologia: Ha una frequenza: 1: 100.000 - 1: 120.000; la tirosine mia è una malattia molto più rara e non è possibile effettuare uno screening neonatale. Presenta un pattern di ereditarietà: autosomica recessiva.
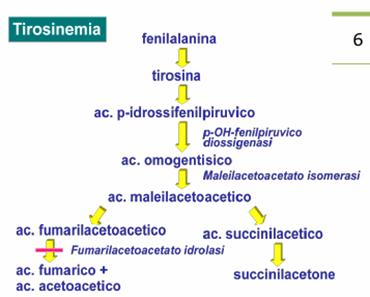 Eziopatogenesi: il difetto di base è a carico della fumaril-aceto-acetato liasi (idrolasi) o deficit di tirosina
amino-trasferasi. La tirosinemia di tipo 1 è causata dal deficit della
fumaril-aceto-acetato idrossilasi mentre la tirosinemia di tipo 2 da un deficit
dell'enzima tirosina amino-trasferasi.
Eziopatogenesi: il difetto di base è a carico della fumaril-aceto-acetato liasi (idrolasi) o deficit di tirosina
amino-trasferasi. La tirosinemia di tipo 1 è causata dal deficit della
fumaril-aceto-acetato idrossilasi mentre la tirosinemia di tipo 2 da un deficit
dell'enzima tirosina amino-trasferasi.
Si accumulano come substrati della via metabolica l'acido succinil-acetico e il succinil-acetone che sono tossici per il fegato. I principali organi colpiti sono il fegato, il rene e i nervi periferici.
![]() La tirosinemia acquisita insorge in seguito ad
una grave insufficienza epatica, scorbuto ed ipertiroidismo.
La tirosinemia acquisita insorge in seguito ad
una grave insufficienza epatica, scorbuto ed ipertiroidismo.
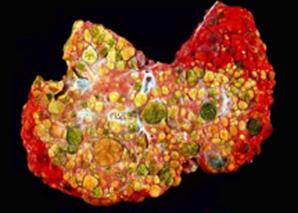 Anatomopatologia: Il fegato di un bambino affetto da tirosinemia va incontro
ad una cirrosi micro o macronodulare. I noduli in verde sono dei noduli cirrotici.
Anatomopatologia: Il fegato di un bambino affetto da tirosinemia va incontro
ad una cirrosi micro o macronodulare. I noduli in verde sono dei noduli cirrotici.
Quadro clinico (tirosinemia di tipo 1)
Stentata crescita;
Insufficienza epatica grave;
Vomito - diarrea;
Ittero;
![]() Sindrome
di Fanconi;
Sindrome
di Fanconi;
Rachitismo;
Neuropatia periferica.
Reperti di laboratorio
aumento della α-feto-proteina;
riduzione dei fattori della coagulazione sintetizzati dal fegato;
aumento dei livelli sierici delle transaminasi;
aumento della concentrazione di bilirubina sierica in presenza di insufficienza epatica.
La diagnosi è confermata dall'aumento dei livelli di succinil-acetone nel sangue e nelle urine. L'analisi del DNA è utile per la diagnosi molecolare pre-natale e per testare gruppi a rischio per specifiche mutazioni.
Terapia: Il trattamento di elezione è il nitisinone (NTBC) che inibisce la degradazione della tirosina a 4-HPPD (4-OH-fenilpiruvato diossigenasi) e previene le crisi epatiche e neurologiche acute.
Il danno epatico precedente al trattamento non è reversibile pertanto i pazienti devono essere monitorati per lo sviluppo di HCC (epatocarcinoma). Il trapianto di fegato costituisce una terapia efficace e riduce il rischio di HCC.
Tirosinemia di tipo 2: Disturbo autosomico recessivo caratterizzato da: ipercheratosi palmare e plantare, ulcere corneali erpetiformi e ritardo mentale.
La diagnosi viene stabilita misurando la concentrazione di tirosina plasmatica e può essere confermata con l'analisi del DNA del gene mutante.
Il trattamento con un ridotto apporto di tirosina e fenilalanina può consentire un miglioramento delle lesioni oculari e cutanee.
Difetti del ciclo dell'urea
![]()
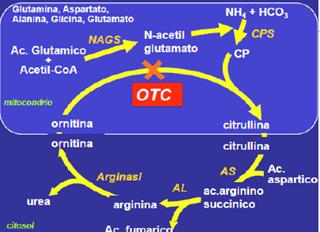 I deficit del ciclo dell'urea possono essere
dovuti a:
I deficit del ciclo dell'urea possono essere
dovuti a:
Deficit di OCT (Ornitina-Carbamil Transferasi);
CPS;
AS (Arginin-Succinico sintetasi);
AL (Arigin-succinico Liasi);
NAGS;
intolleranza alle proteine con lisinuria.
Epidemiologia: La frequenza risulta essere variabile a seconda del deficit enzimatico interessato
1:62'000 - OCT;
- AS;
1:57'000 - AL;
1:70'000 - Arginasi;
1:363'000 - complessiva 1:8'200.
I difetti del ciclo dello'urea costituiscono la più comune causa genetica di iperammonemia neonatale. Considerando la frequenza complessiva i difetti del ciclo dell'urea non sono una patologia alquanto rara.
Per questo motivo, anche i modelli di ereditarietà risultano essere variabili. OCT (Ornitina-Carbamil Transferasi) presenta un pattern di ereditarietà autosomica recessiva o legata all'X recessiva (per questo motivo i maschi sono sempre malati mentre le femmine presentano un quadro sfumato a seconda della Lyonizzazione del cromosoma X).
Quadro clinico: Distinguiamo una presentazione neonatale con iperammoniemia, vomito, tachipnea, torpore, coma, convulsioni, shock, exitus.
Le forme tardive sono caratterizzate da episodi di scompenso ricorrenti. Nei bambini più grandi si presenta con vomito, atassia, confusione mentale, irritabilità, agitazione e aggressività.
Vi è una estrema variabilità di presentazione clinica per soggetti eterozigoti per OCT.
Le indagini di laboratorio mostreranno:
Iperammoniemia;
Ipertransaminasemia;
Iper-aminoacidemia: l'aumento di glutamina è un pattern aspecifico in quanto l'ammonio si lega all'acido glutammico determinando un aumento di glutamina.
L'aumentata escrezione dell'acido orotico orienta verso un difetto delle prime tappe del ciclo dell'urea, un difetto della Carbamil Transferasi.
Il dosaggio dell'ac. orotico urinario mostra un aumento in caso dell'interessamento delle prime tappe del ciclo dell'urea. Un marcato aumento di acido orotico nelle urine consente di fare la diagnosi differenziale tra deficit di OTC e CPS.
Molto importante è il dosaggio degli acidi organici urinari.
Gli esami strumentali effettuati sono:
EEG: frequenti crisi a localizzazione variabile;
Ecoencefalo: nella norma.
Diagnosi: la concentrazione plasmatica di ammoniaca è solitamente superiore a 200 μmoli/L (v.n.<35 μmoli /litro).
Trattamento dell'iperammonemia acuta: Si consiglia la somministrazione e.v. di una corretta quantità di calorie, liquidi ed elettroliti.
Una dose minima di proteine, preferibilmente in forma di amminoacidi essenziali, andrebbe aggiunta per prevenire gli stati catabolici. La somministrazione di arginina è efficace nel trattamento dell'iperammoniemia dovuta a deficit del ciclo dell'urea (eccetto in pz. con deficit di arginasi), perché fornisce al ciclo ornitidina e NAG. Nei pazienti con deficit CPS o OTC è indicata la somministrazione di arginina perché in queste malattie diviene un amminoacido essenziale. La somministrazione di arginina o citrullina è controindicata in pazienti con arginasi.
Nei pazienti con iperammoniemia secondarie ad acidemie organiche il trattamento con arginina non è indicato perché non consente di ottenere alcun effetto benefico.
L'emodialisi anche se rappresenta il sistema più efficace per ridurre l'ammoniaca è una procedura tecnicamente complessa, difficile da eseguire e non sempre disponibile. La dialisi peritoneale è il metodo più rapido e pratico per il trattamento dei pazienti con grave iperammoniemia. Nei pazienti con iperammoniemia da acidemia organica la dialisi peritoneale rimuove efficacemente dal corpo sia gli acidi organici nocivi sia l'ammoniaca.
Può riscontrarsi un intervallo considerevole tra la normalizzazione dell'ammoniaca e il miglioramento delle condizioni neurologiche del paziente. Prima che il neonato recuperi completamente lo stato di allerta sono talvolta necessari diversi giorni.
Acidemie organiche
In questo ambito, bisogna ricordare:
propionico acidemia;
metil-malonico acidemia;
lattico-acidosi.
Metil-malonico acidemia (MMA)
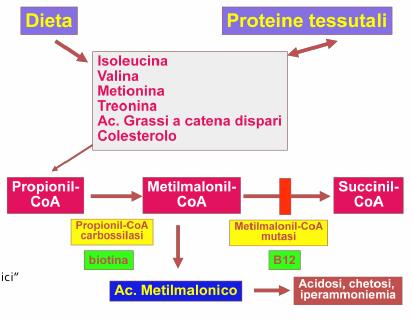 L'acido metil-malonico è un derivato dell'acido
propionico nell'ambito della via catabolica di isoleucina, valina, treonina,
metionina, colesterolo e acidi grassi a catena mista. La conversione dell'acido
metil-malonico ad acido succinico avviene mediante l'azione di due enzimi: la metil-malonil-CoA
racemasi, che forma L-isomero, e la metil-malonil-CoA mutasi che converte
l'acido L-metil-malonico in acido succinico. Quest'ultimo enzima richiede come cofattore
l'adenosil-cobalamina (metabolita della vit. B12).
L'acido metil-malonico è un derivato dell'acido
propionico nell'ambito della via catabolica di isoleucina, valina, treonina,
metionina, colesterolo e acidi grassi a catena mista. La conversione dell'acido
metil-malonico ad acido succinico avviene mediante l'azione di due enzimi: la metil-malonil-CoA
racemasi, che forma L-isomero, e la metil-malonil-CoA mutasi che converte
l'acido L-metil-malonico in acido succinico. Quest'ultimo enzima richiede come cofattore
l'adenosil-cobalamina (metabolita della vit. B12).
![]() Epidemiologia: ha una frequenza di 1:20'000 con ereditarietà autosomica
recessiva.
Epidemiologia: ha una frequenza di 1:20'000 con ereditarietà autosomica
recessiva.
Eziopatogenesi: il difetto di base risiede nella metil-malonil-CoA mutasi, oppure un difetto di sintesi del cofattore vitamina B12 e per questo motivo è importante somministrare vitamina B12 e B6 in questi pazienti.
Quadro clinico: Le forme acute sono caratterizzate da vomito, rifiuto dell'alimentazione, crescita stentata, disidratazione, ipotonia, acidosi, chetosi, iperammoniemia che causa letargia, coma, segni neurologici.
Nelle forme intermittenti o croniche i bambini che sopravvivono al primo attacco possono andare incontro a nuovi episodi metabolici. Tra un attacco e l'altro il paziente presenta ipotonia, disturbi dell'allattamento e deficit della crescita staturo-ponderale. Il QI e lo sviluppo mentale dei pazienti con metil-malonico acidemia possono rientrare nel range della normalità malgrado i ripetuti attacchi.
Reperti di laboratorio: chetosi, acidosi, anemia, neutropenia, trombocitopenia, iperglicemia, iperammonemia ed elevate quantità di acido metil-malonico nei liquidi corporei.
Diagnosi: La diagnosi può essere confermata:
valutando l'incorporazione del proprionato o l'attività della mutasi;
eseguendo il test di complementazione su colture di fibroblasti;
identificando il gene mutante.
Terapia: Il trattamento a lungo termine prevede una dieta a basso contenuto calorico, somministrazione per os di L-carnitina e vitamina B12. Una terapia alcalinizzante cronica è necessaria per correggere l'acidosi cronica, in particolare in prima infanzia.
Propionico acidemia (PA)
Epidemiologia: ha una frequenza di 1:100'000 (più rara della metil-malonico acidemia), con ereditarietà autosomica recessiva.
Eziopatogenesi: il difetto di base risiede nella propionil-CoA-carbossilasi (PCCA, PCCB). L'acido propionico normalmente viene carbossilato dall'enzima mitocondriale propionil-CoA-carbossilasi, che richiede come cofattore la biotina, ad acido metil-malonico.
Quadro clinico: le forme acute sono caratterizzate da letargia, vomito, rifiuto dell'alimentazione, crescita stentata, disidratazione, ipotonia, acidosi, iperammoniemia, coma, segni neurologici.
Le forme intermittenti o croniche si presentano con ritardo mentale, anomalie neurologiche come distonia, tremore, coreo-atetosi, segni piramidali.
Se il bambino sopravvive al primo attacco episodi analoghi tendono a verificarsi a causa di infezioni ricorrenti, costipazione o in seguito ad una dieta ad alto contenuto di proteine.
Reperti di laboratorio: acidosi metabolica, chetosi, neutropenia, trombocitopenia e ipoglicemia. Nei bambini con acidemia propionico si riscontra una marcata elevazione plasmatica e urinaria di acido propionico e acido metil-citrico.
Diagnosi: L'acidemia propionico va in diagnosi differenziale con deficit multipli di carbossilasi. La diagnosi definitiva può essere stabilita misurando l'attività enzimatica su colture di leucociti o fibroblasti.
La diagnosi di laboratorio (MMA e PA)
Acidosi;
Iperammoniemia;
Chetosi;
Test qualitativi urinari (para-nitro-anilina);
Aminoacidemia quantitativa: aumento di GLY, ALA;
Acidi organici urinari (metil-citrato, propionil-glicina, ac. tiglico, 3-OH-propionato, metil-malonato o propionato);
Profilo acil-carnitine plasmatiche (TMS);
Dosaggio enzimatico in fibroblasti;
Analisi molecolare.
Terapia (MMA e PA)
Riduzione dell'apporto dietetico di precursori;
Alimenti speciali e Integratori;
Mantenere un adeguato stato nutrizionale;
Mantenere una normale crescita;
Prevenire il ritardo mentale.
Il trattamento degli attacchi acuti include idratazione, correzione dell'acidosi e miglioramento dello stato catabolico mediante l'apporto calorico con iperalimentazione parenterale. Per i neonati nel corso del primo attacco si consiglia la somministrazione orale di biotina fino alla formulazione della diagnosi.
Malattie del metabolismo dei carboidrati e del metabolismo energetico
In questo ambito, dobbiamo ricordare:
Glicogenosi;
Galattosemia;
intolleranza ereditaria al fruttosio.
Glicogenosi
Le glicogenosi sono un gruppo di malattie metaboliche rare che colpiscono un bambino su 12.000 nati. Sono dovute alla carenza o al deficit funzionale di uno degli enzimi coinvolti nel metabolismo del glicogeno, il polisaccaride che funge da deposito e da riserva per le molecole di glucosio, che l'organismo utilizza prontamente in caso di bisogno di energia. Un accumulo di glicogeno nei tessuti (fegato, muscoli, rene, cervello) provoca gravi alterazioni organiche. Le persone affette da glicogenosi, non potendo utilizzare i propri depositi di zuccheri, sono costrette a mangiare continuamente sia di giorno che di notte per evitare di cadere in ipoglicemia, con possibile insorgenza di convulsioni e coma, pertanto richiedono una continua e attenta sorveglianza.
I difetti enzimatici alla base delle glicogenosi sono causati da errori del genoma, che si trasmettono per via ereditaria come fenotipo autosomico recessivo, tranne il tipo VIII, che si eredita come fenotipo recessivo legato al cromosoma X.
Classificazione: Esistono più di 12 forme di glicogenosi.
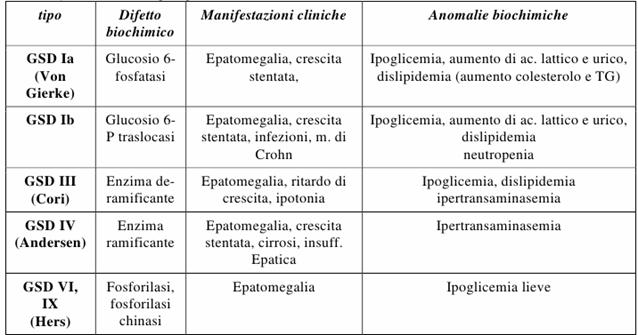
Figura : Classificazione delle glicogenosi.
GSD tipo I
È una forma severa che colpisce 1/centomila abitanti, l'enzima mancante è il G6P-fosfatasi presente solo nel fegato; i soggetti affetti da questa patologia non possono resistere al digiuno, andando incontro velocemente ad una crisi ipoglicemica. il G6P-fosfatasi è l'unico enzima in grado di convertire il glicogeno in glucosio, ed è presente all'interno del reticolo endoplasmatico. Si trova solo nel fegato e in piccole percentuali nel rene, idrolizza anche il carbamil fosfato e il pirofosfato, si forma nel citosol e un trasportatore lo veicola al RE. Tuttavia in alcune glicogenosi di tipo I il G6P-fosfatasi può essere presente ma viene a mancare il trasportatore di questo che lo veicola al RE.
I sintomi che caratterizzano questo tipo di glicogenosi sono: ipoglicemia a digiuno, epatomegalia (dovuta ad un aumento del glicogeno nel fegato), ritardata crescita nell'adolescenza, iperlipidemia con possibile steatosi, aumento dei corpi chetonici e un aumento degli acidi grassi in circolo (perchè l'ipoglicemia induce iperinsulinemia). Esistono forme diverse di glicogenosi di tipo I:
Ia dove è assente l'unità catalitica di G6P-fosfatasi: la glucosio 6 fosfatasi è presente all'interno dei microsomi con la regione catalitica all'interno del microsoma. Il deficit di glucosio-6-fosfatasi determina ipoglicemia da mancata fuoriuscita di glucosio;
Ia SP dove manca una proteina che stabilizza l'enzima;
Ib manca il trasportatore per il RE: 'enzima glucosio 6-traslocasi, deficitario nella GSD 1b, trasloca il glucosio-6P all'interno dell'organo del Golgi. Nella GSD 1b non avviene l'interazione tra il glucosio-6P con l'enzima glucosio 6 fosfatasi;
Ic manca il trasportatore di Pi e PP;
Id manca il trasportatore del glucosio dal citosol al reticolo.
La GSD di tipo I si trasmette con modalità autosomica recessiva.
Esordio: La patologia può esordire nel periodo neonatale con ipoglicemia e acidosi lattica. La presentazione più comune è tra i 3-4 mesi di età con epatosplenomegalia e/o convulsioni ipoglicemiche. Può riscontrarsi diarrea intermittente.
Semeiotica: I bambini colpiti presentano la tipica "faccia a bambola", arti brevi, bassa statura e addome prominente. Questi pazienti presentano un'immunodeficienza secondaria: infezioni ricorrenti dovute alla neutropenia e alla compromissione della funzione neutrofila, granulomi simili a quelli della malattia di Crohn, ulcerazioni della mucosa orale e intestinale e MICI.
Le caratteristiche biochimiche sono: ipoglicemia, acidosi lattica, iperuricemia e iperlipidemia.
Complicanze: Nella totalità delle pazienti di sesso femminile è presente ovaio policistico. I sintomi della gotta si presentano in pubertà. Le anomalie lipidiche comportano un aumentato rischio di pancreatite. Le fratture ossee sono frequenti per la presenza di osteopenia. Sviluppano intorno alla seconda.-terza decade di vita adenomi epatici soggetti ad emorragie che in alcuni casi diventano maligni. Altra complicanza è la malattia renale.
Prognosi: La diagnosi e il trattamento precoce hanno migliorato la prognosi di questi pazienti ma la malattia renale e gli adenomi epatici sono complicanze gravi.
GSD tipo II
L'enzima mancante è l'α 1-4 glucosidasi, questo enzima non è specifico per il glicogeno ma idrolizza anche i legami del maltosio (disaccaride costituito da due molecole di glucosio con legame α 1-4 glucosidico) e tutti gli altri materiali aventi questo tipo di legame. L'accumulo si presenta a carico di tutti i tessuti ma soprattutto a livello del fegato e del muscolo scheletrico. Non si riscontrano alterazioni dell'omeostasi glicemica. Esiste una forma infantile severa caratterizzata da cardiomegalia, ipotonia muscolare ed epatomegalia, in questo caso il decesso del paziente si riscontrerà entro i primi 2-3 anni di vita.
GSD tipo III
La malattia colpisce sia il fegato che i muscoli ed è definita GSD IIIa mentre nel 15% dei casi colpisce solo il fegato ed è definita GSD IIIb. Pazienti affetti da GSD III presentano ipoglicemia facilmente controllabile, una dislipidemia non così marcata come quella della GSD1 mentre l'ipertransaminasemia è 10 volte più severa: il fegato va incontro a fibrosi. Inoltre nella GSD IIIa è presente anche un interessamento muscolare con un aumento del CPK e una ipertransaminasemia anche di tipo muscolare.
L'enzima mancante è l'enzima deramificante, quindi il glicogeno riesce ad essere degradato solo parzialmente e il quantitativo di glucosio liberato sarà ridotto. Si instaura un aumento di glicogeno epatico e muscolare, caratterizzato da numerose ramificazioni e catene laterali molto corte.
Presentazione clinica: Condizioni cliniche generali scadute, facies sofferente, colorito pallido, occhi alonati, capelli fini e radi. Epatomegalia (fegato di consistenza ridotta, palpabile in fossa iliaca dx). Il fegato non è di consistenza dura come quando il paziente presenta la cirrosi mentre in corso di malattie metaboliche è talmente grande e soffice che a volte per errore si crede di non palparlo. È presente irritabilità.
Diagnostica: Profilo glicemico di 24 ore: episodi di ipoglicemia (soprattutto dopo digiuni prolungati), grave ipetransaminasemia e CPK aumentato; può non essere aumentato nella fase iniziale della malattia in quanto in questo primo periodo può non essere presente l'interessamento muscolare.
 L'Eco addome mostra: epatomegalia (dx 150 mm),
aspetto steatosico del fegato. Il test da carico con il glucagone dimostra una
insensibilità nella liberazione di glucosio dal glicogeno epatico dopo il
pasto. Questo quadro è compatibile con il deficit dell'enzima deramificante
come avviene nella glicogenosi III. Il test da carico con il glucagone permette
di distinguere le forme di malattia in cui è possibile mettere in circolo il
glucosio. Un paziente che presenta un deficit dell'enzima de ramificante presenta
un appiattimento della curva rispetto al paziente normale in cui si ha il
rilascio di glucosio a partire dal glicogeno epatico.
L'Eco addome mostra: epatomegalia (dx 150 mm),
aspetto steatosico del fegato. Il test da carico con il glucagone dimostra una
insensibilità nella liberazione di glucosio dal glicogeno epatico dopo il
pasto. Questo quadro è compatibile con il deficit dell'enzima deramificante
come avviene nella glicogenosi III. Il test da carico con il glucagone permette
di distinguere le forme di malattia in cui è possibile mettere in circolo il
glucosio. Un paziente che presenta un deficit dell'enzima de ramificante presenta
un appiattimento della curva rispetto al paziente normale in cui si ha il
rilascio di glucosio a partire dal glicogeno epatico.
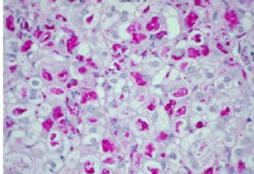
All'agobiopsia epatica, la struttura globulare è conservata, epatociti di taglia ampia, con membrana cellulare ispessita e rigida. Citoplasma rigonfio, rarefatto e roseo. Gli epatociti appaiono fortemente colorati al periodico acid Schiff (PAS) per l'eccesso di glicogeno che appare digerito dalla diastasi. Moderata fibrosi portale con formazione di sottili setti e fibrosi pericellulare. Dosaggio glicogeno intra-eritrocitario: aumentato (v.n.< 100 mg/g di Hb).
![]() È possibile poi effettuare un dosaggio eritrocitario
dell'attività dell'enzima e analisi molecolare.
È possibile poi effettuare un dosaggio eritrocitario
dell'attività dell'enzima e analisi molecolare.
Complicanze: In pazienti con cirrosi epatica progressiva sono stati segnalati HCC. L'ipertrofia ventricolare è un riscontro frequente, anche l'ovaio policistico è comune. La somministrazione di glucagone 2 ore dopo un pasto a base di carboidrati comporta un normale aumento di glucosio nel sangue. Dopo una notte di digiuno è possibile che il glucagone non determini alcun aumento dei livelli sierici ematici di glucosio.
GSD tipo IV
È tra le più rare di tutte le glicogenosi, l'enzima mancante è l'enzima ramificante (glucosil 4-6 transferasi), l'assenza di questo comporta una sintesi anomala di glicogeno con scarse ramificazioni e quindi poco solubile. Le manifestazioni cliniche sono: epatomegalia, cirrosi con ipertensione portale, ipotono muscolare e morte nei primi 2-3 anni di vita. È una patologia multisistemica. Questi pazienti presentano noduli precoci e vanno incontro a cirrosi precoce e trasformazione ad HCC. E' stata valutata anche una forma muscolare che esordisce alla nascita con grave ipotonia, atrofia muscolare e coinvolgimento neuronale.
Altri tipi di glicogenosi
Vengono riportati gli altri tipi di glicogenosi:
GSD tipo V: deficit di fosforilasi muscolare, malattia di McArdle si manifesta in individui nella seconda-terza decade, in soggetti che presentano una storia di mioglobinuria, dolori e crampi muscolari;
GSD tipo VI: deficit di fosforilasi epatica, malattia di Hers può essere X-Linked: con un deficit dell'attività fosforil-chinasica del fegato oppure Autosomica con deficit dell'attività fosforil-chinasica a carico di fegato e muscolo o con deficit dell'attività fosforilasica del fegato;
GSD tipo VII: deficit di fosfofruttochinasi muscolare, malattia di Tarui, il compito di questo enzima è quello di convertire il F6P in F1,6BP. È un enzima chiave nella regolazione della glicolisi. Manifestazioni cliniche sono: affaticamento muscolare e intolleranza all'esercizio fisico;
GSD tipo VIII: caratterizzata dalla presenza dell'enzima fosforilasi epatica ma in forma inattiva. Manifestazioni cliniche sono: epatomegalia presente dalla prima infanzia e progressiva cerebropatia degenerativa.
I pazienti affetti da GSD VI e IX presentano forme lievi con modeste epatomegalia ed ipoglicemia che tendono a risolversi con il passare degli anni e quindi presenti solo in età pediatrica. Nella GSD di tipo VI una dieta a elevato numero di carboidrati e pasti frequenti sono efficaci nel prevenire l'ipoglicemia. La diagnosi richiede il dosaggio enzimatico su biopsia epatica.
Intolleranza ereditaria al fruttosio (IEF)
Epidemiologia: Frequenza 1:20'000 - 1:40'000
Eziopatogenesi: presenta una ereditarietà autosomica recessiva. Il difetto di base è l'aldolasi B (frutto-aldolasi B), che mostra una attività ridotta in fegato, reni ed intestino.
Patogenesi: tossicità di F1P (fruttosio 1 fosfato) o riduzione di ATP. Se l'assunzione di fruttosio è continua si verificano ricorrenti episodi ipoglicemici e l'insufficienza epatica e renale progrediscono con esiti potenzialmente fatali.
Quadro clinico: danno epatico, ipertransaminasemia, steatosi epatica, ittero, epatomegalia, letargia, convulsioni; rifiuto di cibi contenenti fruttosio; ipoglicemia (inibizione della gluconeogenesi e della fosforilasi) vomito, stentata crescita.
Reperti di laboratorio: tempo di coagulazione aumentato, ipoalbuminemia, aumento livelli bilirubina e transaminasi, disfunzione tubulare prossimale.
Diagnosi: Deve essere sospettata per la presenza di una sostanza riducente nelle urine nel corso di un episodio ipoglicemico.
Galattosemia
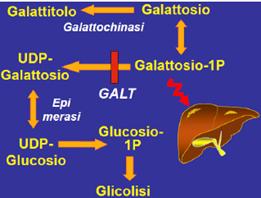 Epidemiologia: Frequenza di 1:40'000 - 1:80'000.
Epidemiologia: Frequenza di 1:40'000 - 1:80'000.
Eziopatogenesi: presenta una ereditarietà autosomica recessiva. Il difetto di base risiede nella galattosio-1P-uridil-transferasi (GALT). Esistono 2 forme diverse del disturbo: la prima è caratterizzata dal deficit completo (o quasi) dell'enzima mentre la seconda da un deficit parziale. In assenza del GALT il galattosio 1-P si accumula nei reni, nel fegato e nel cervello.
![]() Quadro clinico: Sepsi neonatale (E.Coli). La sepsi da E.coli è frequente
in questi pazienti in quanto probabilmente il batterio cresce meglio in presenza
di galattosio.
Quadro clinico: Sepsi neonatale (E.Coli). La sepsi da E.coli è frequente
in questi pazienti in quanto probabilmente il batterio cresce meglio in presenza
di galattosio.
Vi è un grave danno epatico, con necrosi epatocellulare con ittero. La galattosemia è una delle cause di colestasi del neonato. La mortalità nei primi giorni di vita in seguito ad insufficienza epatica e renale è elevata.
Altre segni e sintomi sono: crescita stentata, cataratta, danno renale, diarrea, vomito, anemia. Il deficit parziale è in genere asintomatico.
Diagnosi: Il deficit parziale, in quanto asintomatico, è diagnosticato mediante lo screening neonatale. La diagnosi viene formulata in seguito alla dimostrazione di sostanze riducenti in diversi campioni di urine prelevati quando il paziente segue ancora la dieta con il latte umano, latte vaccino o latte in polvere contenenti lattosio. La sostanza riducente individuata con il clini test può essere identificata mediante cromatografia o test enzimatico specifico per il galattosio.
Malattie da accumulo lisosomiale
Le malattie da accumulo lisosomiale o (LSD) acronimo dall'inglese Lysosomial Storage Disease sono un'eterogenea famiglia di patologie, circa 50, dovute a diversi tipi di difetti genetici, accomunate dalla caratteristica di determinare un accumulo di metaboliti o sostanze nei lisosomi con conseguente perdita di funzionalità cellulare. Le cause di queste patologie sono sempre da ricondurre ad un'anomalia genetica, trasmessa con modalità autosomica recessiva oppure di tipo X-linked recessivo, come la malattia di Fabry e la sindrome di Hunter (MPS II).
Cenni storici: Una delle prima individuate è stata la malattia di Tay-Sachs nel 1881, nel 1882 seguita dalla malattia di Gaucher. Solo verso la fine degli anni sessanta de Duve e coll., utilizzando tecniche di frazionamento cellulare, studi citologici e analisi biochimiche, identificarono e caratterizzano il lisosoma, riconoscendone la funzione di organello cellulare responsabile della digestione intracellulare e del riciclaggio delle macromolecole. Ciò ha permesso la comprensione delle basi fisiologiche delle malattie da accumulo lisosomiale.
La malattia di Pompe è stata la prima ad essere riconosciuta come una LSD nel 1963 in seguito agli studi di L. Hers, il quale scoprì che essa è dovuta ad un deficit di α-glucosidasi. Lo stesso ricercatore intuì che anche le MPS (mucopolisaccaridosi) potevano essere dovute a carenze enzimatiche.
Eziologia ed epidemiologia: Di solito queste patologie sono dovute a disfunzioni lisosomiali da carenza di un singolo enzima necessario per il metabolismo di lipidi, glicoproteine (proteine contenenti zucchero) o dei mucopolisaccaridi. L'incidenza di ciascuna patologia, presa singolarmente, è inferiore a 1:100.000; complessivamente, però, questo gruppo di malattie ha un'incidenza di 1:5000 - 1:10.000.
Patogenesi: Il lisosoma ha un ruolo chiave nel metabolismo delle sostanze endogene, che se accumulate all'interno della cellula sono tossiche. Quando gli enzimi in esso contenuti e confinati sono carenti o assenti il metabolismo dei prodotti di rifiuto del metabolismo cellulare non avviene con efficienza e le sostanze, accumulandosi all'interno della cellula, determinano tossicità fino alla morte cellulare. In altri termini, quando i lisosomi non funzionano normalmente, i prodotti in eccesso destinati alla demolizione enzimatica da parte degli enzimi contenuti nei lisosomi si accumulano nella cellula.
Tutte le malattie lisosomiali hanno origine da un accumulo anomalo di sostanze all'interno dei lisosomi. Esse sono dovute a deficit genetici ereditati dai genitori, deficit che determinano una carenza/assenza enzimatica specifica per ogni singola patologia. Queste patologie hanno un esordio infantile che va da pochi mesi a pochi anni dalla nascita. Molti bambini affetti muoino dopo pochi anni dalla presentazione della sintomatologia, con sintomi ingravescenti e spesso devastanti. I sintomi delle varie patologie variano tra loro a seconda dello specifico disturbo, e di variabili come l'età di insorgenza; la gravità delle manifestazioni è variabile.
Classificazione
Mucopolisaccaridosi:
m. di Hurler, m. di Scheie (MPS1);
m. di Hunter (MPS II);
m. di San Filippo (MPS III);
m. di Morquio (MPS IV);
m. di Scheie (MPS VI);
m. di Sly (MPS VII)
Gangliosidosi:
m. di Tay-Sachs;
Gangliosidosi GM1, etc
Sfingolipidosi:
m. di Gaucher;
m. di Niemann-Pick.
Sono più rare: la leucodistrofia metacromatica ed il m. di Krabbe, etc..
Oligosaccaridosi:
Fucosidosi;
Mannosidosi;
Sialidosi, etc
Malattie da accumulo misto.
Manifestazioni cliniche
Coinvolgimento SNC;
Dismorfismi facciali (faccia "a mascherone") dovuta a displasia ossea;
Visceromegalia;
Segni ematologici;
Segni oculari (cataratta).
Le mucopolisaccaridosi sono malattie ereditarie e progressive causate da mutazioni di geni che codificano per enzimi lisosomiali necessari per degradare i glicosaminoglicani.
Malattia di Hurler o MPS I
La malattia di Pfaudler-Hurler o gargoilismo (per richiamare l'aspetto che ricorda un gargoyle) è una rara patologia genetica (carattere recessivo) caratterizzata da nanismo disarmonico (accorciamento del collo e del tronco con addome prominente per epatosplenomegalia), ritardo psichico e alterazioni corneali e cutanee. Questa malattia è una mucopolisaccaridosi e fa parte dell'eterogeneo gruppo delle malattie da accumulo lisosomiale.
L'incidenza della malattia risulta un caso su 100.000 persone.
L'aspetto di questi pazienti è particolarmente scimmiesco: fronte prominente, radice del naso infossata, cute ispessita, labbra grosse, lingua protrudente, cornee opache, ritardo mentale. Sono stati riscontrati anche difetti al cuore e alle valvole cardiache, ritardo mentale ed epatosplenomegalia. La patologia è caratterizzata anche da un disturbo metabolico connettivale con iperproduzione di alcuni mucopolisaccaridi, dovuto al deficit dell'enzima α-L-ialuronidasi. Se i difetti cardiaci sono presenti la morte può avvenire nell'adolescenza per la cardiopatia.
Clinica: Peso, altezza, P/A, circ. cranica sono superiori al 95° percentile. Facies grossolana: tratti grossolani, fronte prominente, naso insellato (una depressione del naso), macroglossia, ipertrofia gengivale, mento particolare. Addome globoso con ernia ombelicale, visceromegalia. Limitazioni articolari e torace carenato. Lieve ritardo mentale. Infezioni ricorrenti delle vie respiratorie superiori, otiti, stenosi coronarica e morte prematura del paziente
Va in diagnosi differenziale con l'acondroplasia.
Tutti questi segni consentono di fare diagnosi di malattia da accumulo lisosomiale.
La MPS II è una malattia X-linked e si presenta quasi esclusivamente nei maschi. Nella MPS III è presente un grave coinvolgimento del SNC lentamente progressivo e lievi sintomi somatici. Nelle MPS IV e VI non è presente il ritardo mentale mentre si ritrovano opacità corneali, disostosi multiple. I pazienti con MPS di tipo VI presentano anche visceromegalia.
M. di Gaucher
La malattia di Gaucher è una lipidosi multisistemica caratterizzata da problemi ematologici, organomegalia e coinvolgimento scheletrico. È il risultato del deficit dell'idrolasi lisosomiale che comporta un accumulo di substrati glicolipidici non degradati.
![]()
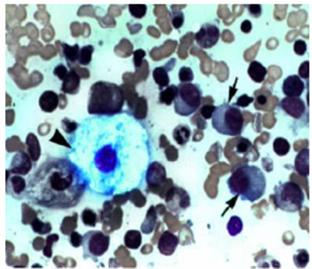 Questi pazienti presentano una
epato-splenomegalia.
Questi pazienti presentano una
epato-splenomegalia.
Nell'immagine è presente la cellula di Gaucher con i vacuoli che contengono materiale di accumulo.. Il tipo 1 è più frequente, questi pazienti non presentano ritardo mentale e possono vivere fino ad 80 anni.
Diagnosi di Malattie da accumulo
E' importante valutare la facies grossolana, il ritardo mentale, la cataratta, i segni ematologici ecc.
Le indagini di I livello comprendono:
screening urinari (MPS, oligosaccaridi). Sono test qualitativi che utilizzano reazioni colorimetriche e/o cromatografie;
Rx scheletro;
striscio periferico (cellule di accumulo);
visita oculistica (lampada a fessura, fundus).
Sono indagini di II livello:
identificazione di metaboliti urinari con cromatografie;
dosaggi enzimatici in plasma, leucociti o fibroblasti fatti crescere in coltura. Si può valutare l'α-gluconidasi, la glucoronato-fosfatasi, la lattosio-6-solfatasi, l'N-acetil-galattosio-4-solfatasi, la β-glucuronidasi ecc;
analisi molecolare.
Malattie perossisomiali
Sono caratterizzate da gravi quadri di ipotonia e spesso sono letali. Questi difetti geneticamente determinati sono causati o dall'impossibilità di formare oppure di mantenere il perossisoma o dal deficit di funzione di un singolo enzima normalmente localizzato in questo organello.
I difetti perossisomiali sono suddivisi in due categorie principali: difetti di biogenesi perossisomiale e difetto che coinvolge la singola proteina perossisomiale.
Difetti della biosintesi del colesterolo
Sono malattie associate alla riduzione di colesterolo. La sindrome di Smith-Lemli-Opitz (o sindrome SLO) è una sindrome autosomica recessiva, che causa malformazioni congenite multiple e ritardo mentale. La malattia si può manifestare con fenotipi più o meno gravi, tanto che prima veniva distinta in tipo 1 più lieve e 2 più grave. La patologia è causata dalla mutazione di un gene sul braccio q del cromosoma 11, che codifica per l'enzima 7-deidrocolesterolo reduttasi.
Caratteristica degli individui affetti dalla malattia è la scarsa presenza di colesterolo e l'alta quantità di 7-deidrocolesterolo.
Manifestazioni cliniche: Letalità perinatale, ritardo mentale, ritardo di crescita, dismorfismi cranio-facciali, anomalie degli arti, anomalie dei genitali, malformazioni interne, cataratta, anomalie strutturali cerebrali.
Diagnosi delle Malattie Metaboliche
Al sospetto diagnostico seguono i test di laboratorio di 1° livello, con analisi dei metaboliti, dosaggio enzimatico e infine analisi molecolare.
Le indagini iniziali nel sospetto di malattia metabolica vengono effettuate su campioni di urine, sangue.

Figura : Indagini di base per la diagnosi degli errori congeniti del metabolismo.
L'uso di test di laboratorio di primo livello può consentire un inquadramento sufficientemente preciso della malattia o del gruppo di malattie metaboliche. È importante conservare post-mortem tutte le indagini effettuate al bambino per la programmazione di gravidanze future.
Lo screening neonatale viene effettuato mediante prelievo di sangue da tallone, successivamente viene messo su carta bibula e viene spedito al centro di screening dove viene analizzato. Se il test risulta positivo, e quindi si ritrova un metabolita aumentato, si procede con un'ulteriore indagine (ad esempio amminoacidemia). Se la seconda indagine conferma la malattia metabolica il paziente viene inviato in un centro di follow-up.
Terapia delle Malattie Metaboliche
I principi generali sono:
riduzione dell'accumulo di metaboliti dannosi;
supplementazione di metaboliti carenti;
detossificazione di metaboliti dannosi;
stimolazione di vie metaboliche alternative;
supplementazione di cofattori;
inibizione della produzione di metaboliti tossici (es. succinil-acetone nella tirosinemia);
aumento della disponibilità dell'enzima (terapia sostitutiva enzimatica nella malattia di Gaucher).
La dieta dovrà prevedere un ridotto apporto di specifici componenti, alimenti speciali, integratori,vitamine, frequenza dei pasti stabilita.
La terapia enzimatica sostitutiva può essere utilizzata nella malattia di Gaucher:. Altre terapie in corso di sperimentazione per: MPS 1 (m. di Hurler - m. di Scheie); MPS 2 (m. di Hunter); MPS 6 (m. di Maroteaux-Lamy); glicogenosi 2 (m. di Pompe); M. di Niemann-Pick B.
La terapia dello scompenso acuto prevede:
Eliminare dalla dieta i precursori dei metaboliti tossici;
Fornire alimentazione normo-ipercalorica (120-150 kcal/kg/die per bambini, 80-100 per ragazzi, 40-50 per adulti) con integratori calorici e formule prive di precursori (gavage, glucosata, lipidi iv);
Fornire adeguato apporto di liquidi (gavage, iv);
Correggere squilibri associati (disidratazione, acidosi, elettroliti).
Tra gli approcci terapeutici alternativi vi è soprattutto il trapianto, indicato per:
Trapianto di midollo per mal. Lisosomiali;
Trapianto di fegato per aminoacidopatie, mal. del metabolismo dei carboidrati;
Trapianto di cellule epatiche per mal. del metabolismo dei carboidrati.
Infine, grande entusiasmo desta la terapia genica.
|
| Appunti su: https:wwwappuntimaniacomuniversitamedicinamalattie-metaboliche-bambino45php, malattie metaboliche bambino, dieta per tirosinemia di tipo 2, acido succinilacetico, deficit OTC, |
|
| Appunti Bellezza |  |
| Tesine Bambini |  |
| Lezioni Nutrizione |  |