|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 4486 | Gradito: |
Leggi anche appunti:Anticolinesterasicidi: Marco Napoli Anticolinesterasici Tesi - Tiroidectomia mini-invasiva video- assistita e Tiroidectomia trans-ascellare robot- assistitaIntroduzione Estensione e profonditÀ delle ustioniESTENSIONE E PROFONDITÀ DELLE USTIONI L'estensione e la profondità delle |
 |
 |
LEUCEMIA IN ETA' PEDIATRICA
La leucemia è una malattia sistemica legata alla proliferazione clonale ed incontrollata di cellule emopoietiche immature. Rappresenta il tumore più frequente in età pediatrica (1/3 di tutte le neoplasie infantili). Il picco di incidenza si ha tra i 4-5 anni colpendo maggiormente i maschi.
Le leucemie vengono classificate in base a:
cellula da cui hanno origine: linfoide nell'85% delle leucemie in età pediatrica e mieloide;
tempo di progressione: acute e croniche.
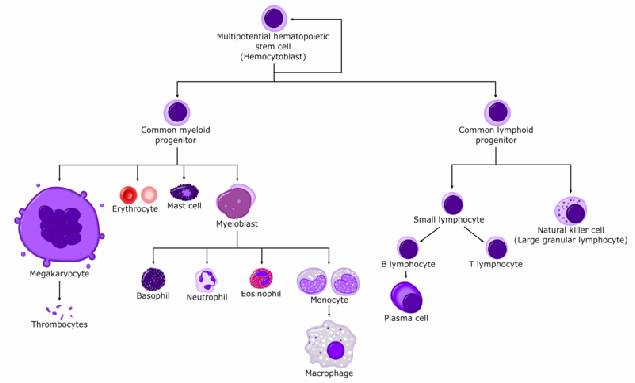
Figura : Linee cellulari della serie mieloide.
Si ricordi invece che nell'adulto le leucemie hanno origine da un precursore mieloide e che, in funzione del suo grado di differenziazione, si effettua la stadiazione.
Molto spesso, le leucemie si presentano in associazione con altre sindromi, quali quella di Down, di Klinefelter, ecc Molto probabilmente, l'associazione con altre sindromi è dovuta a difetti nella risposta al danno delle cellule, che ovviamente tendono ad accumulare un numero maggiore di mutazioni, fino a sfociare nella genesi di un clone neoplastico. La presenza di sindromi rappresenta il fattore di rischio individuale.
Fattori di rischio socio-ambientali sono invece:
età dei genitori elevata;
esposizione dei genitori a particolari ambienti quali industrie chimiche e nucleari, a piombo, petrolio, industria alimentare, raggi X, fumo di tabacco, gas anestetici;
esposizione del piccolo paziente a radiazioni ionizzanti, campi elettromagnetici, cloramfenicolo.
Leucemia linfoblastica acuta
La leucemia linfoblastica acuta è una malattia linfopriliferativa originata da un primitivo disordine dell'emopoiesi, che si manifesta con una proliferazione neoplastica, clonale, automantenuta, di precursori della linea linfoide a livello midollare.
La leucemia linfoblastica acuta (LLA) è la più frequente neoplasia dell'età pediatrica, costituendo più di un terzo di tutti i tumori dell'infanzia e circa l'80% di tutti i casi di leucemia acuta. L'incidenza annuale è di circa 39 nuovi casi per ogni milione di soggetti di età inferiore a 15 anni. Il picco di incidenza è tra i 2-5 anni di età, con un rapporto M/F di 1,2-1,4/1.
La presentazione clinica della LLA è dovuta alla proliferazione degli elementi blastici a livello del midollo. Tali elementi blastici agiscono su più livelli a livello midollare. Innanzitutto si sostituiscono alle cellule normali, causando insufficienza midollare, che clinicamente si traduce in anemia, neutropenia e piastrinopenia. I blasti liberano sostanze attive, determinando febbre e CID (coaugulazione intravasale diffusa) e possono infiltrare altri parenchimi, dando luogo a organomegalia e deficit funzionali. Circa il 90% dei soggetti manifesta astenia, anoressia, pallore ed anemia, un 70% infezione e/o febbre, un 50% emorragie da piastinopenia e un 75% dolori ossei.
Nell'80% dei casi l'anemia, con Hb inferiore ai 10 g/dL, è di tipo normocromica e normocitica, con una quota di reticolociti bassa. I segni dell'anemia sono pallore cutaneo e mucoso, astenia e facile affaticabilità, dispnea da sforzo ed a riposo, tachicardia, edemi declivi e sudorazione abbondante.
La piastrinopenia è responsabile di emorragie cutanee con petecchie, ecchimosi ed ematomi, di emorragie mucose con epistassi, gengivorragie, ematemesi, melena e rettorragia, emorragie parenchimali con ematuria micro e macroscopica, emorragia cerebrale.
La neutropenia è responsabile di infezioni batteriche localizzate o disseminate, e di infezioni fungine al cavo orale, alla vulva e al retto.
L'infiltrazione del clone neoplastico dei parenchimi è responsabile di:
linfoadenomegalia, nel 70% dei casi;
epatomegalia, nel 64% dei casi;
splenomagalia nel 59% dei casi;
dolori ossei, con zoppia o rifiuto alla deambulazione nel 25% dei casi.
In alcuni casi si verificano localizzazioni a livello di rene, mediastino, testicolo e occhio, raramente ad intestino, cute, polmone. In un 3% dei casi vi è una localizzazione a livello del SNC dando quadri di meningosi e sintomi dovuti all'infiltrazione parenchimale. La meningosi esordisce con una sitomatologia meningea e papilledema. All'esame del liquor si avrà una pressione aumentata, un aumento del contenuto in proteine e una riduzione del glucosio, inoltre alla citocentrifuga si reperta la presenza di blasti. L'infiltrazione parenchimale esordisce con emiparesi, paralisi dei nervi cranici e convulsioni. A seconda dell'area del SNC che risulta essere infiltrata avremo:
sindrome cerebellare con atassia, discinesie, ipotonia e iperreflessia;
sindrome ipotalamica, con bulimia, aumento di peso, irsutismo, diabete insipido;
sindrome spinale, con parestesie, sciatalgia, incontinenza sfinterica.
La sintomatologia dell'interessamento del SNC è dovuto anche all'effetto massa, dando il quadro di leucostasi cerebrale. Infatti la massa determina un pericoloso rallentamento della circolazione del sangue per la presenza di un gran numero di blasti leucemici (oltre i 100000/μL).
Le complicanze vita-limitanti alla diagnosi sono:
compromissione dello stato ematologico: anemia grave, infezioni severe, emorragie, trombosi, leucostasi a livello di vari organi ed apparati;
infiltrazione diretta di organi da parte di cellule leucemiche;
sindrome da lisi tumorale, nei casi di iperleucocitosi e di importante carico leucemico.
La sindrome da lisi tumorale è dovuta all'enorme lisi blastica con rilascio in circolo di composti tossici per determinati organi, quali il cuore. La lisi, a livello laboratoristico determina iperuricemia, iperkaliemia, iperfosforemia, ipomagnesemia e iper o ipocalcemia.
Passiamo adesso a descrivere l'iter diagnostico. Il tutto parte dall'esame obiettivo che sottolinerà la presenza di organomegalie, linfonodi ingrossati, emorragie, lesioni cutanee, segni neurologici e segni di infezione. A questo punto si eseguono gli esami emocromocitometrici che mostreranno la presenza di anemia, leucopenia/leucocitosi, piastrinopenia e blastosi. Il sangue periferico presentera una bassa conta di globuli rossi, emoglobina e piastrine, un aumento dei globuli bianchi e la presenza di blasti. Il prossimo passo è quello di un esame del sangue midollare, che mostrerà un'estesa infiltrazione blastica più o meno completa in oltre il 25% dei casi. È necessario effettuare le analisi sia sul sangue periferico che midollare per poter così ottenere studi morfologici, immunologici, citogenetici, biochimici e biologico-molecolari per una precisa caratterizzazione della forma leucemica. Le sedi elettive su cui praticare l'aspirato midollare variano in base all'età. La cresta iliaca può essere utilizzata a qualunque età, mentre il femore e la tibia sono le sedi di elezione nei bambini con età compresa tra 0-2 anni. Oltre i due anni si preferisce il processo spinoso vertebrale, mente oltre i 6 anni lo sterno.
Una volta prelevato il campione si procede ad effettuare la visualizzazione al microscopio, ed è proprio in base a questa che si attua la classificazione FAB. Distinguiamo tre stadi, L1, L2, L3.
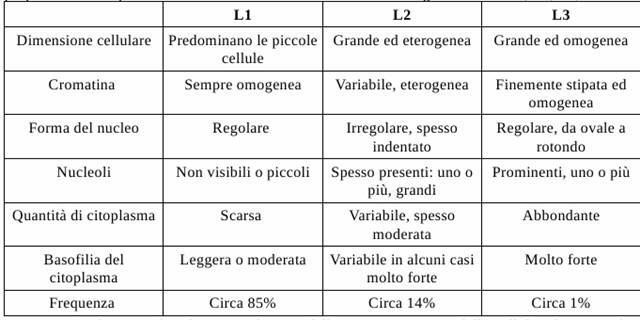
Figura : Classificazione FAB della leucemia linfoblastica acuta.
![]()
 Poi si passa ad una analisi di tipo qualitativo
dell'assetto enzimatico delle cellule, che rispecchia anche il grado di
differenziazione dei blasti. Le principali reazioni citochimiche sono riportate
nella tabella.
Poi si passa ad una analisi di tipo qualitativo
dell'assetto enzimatico delle cellule, che rispecchia anche il grado di
differenziazione dei blasti. Le principali reazioni citochimiche sono riportate
nella tabella.
![]()
 Infine, si effettua la classificazione immunologica, con
la ricerca di particolari antigeni. Questo è molto utile soprattutto per la
terapia e la prognosi.
Infine, si effettua la classificazione immunologica, con
la ricerca di particolari antigeni. Questo è molto utile soprattutto per la
terapia e la prognosi.
La conoscenza di tutti questi fattori è molto utile nel valutare la prognosi del paziente. I fattori prognostici sono parametrici clinici e/o biologici presenti nel paziente alla diagnosi e correlati all'outcome che vengono utilizzati per stratificare i pazienti in fasce di rischio al fine di modulare l'aggressività del trattamento. I fattori prognostici sono suddivisi in favorevoli e sfavorevoli e sono riportati nella tabella.

Figura : Fattori prognostici.
I soggetti che presentano fattori prognostici positivi hanno una sopravvivenza a 5 anni superiore all'84%.
La terapia si articola in diverse fasi. L'induzione ha il fine di indurre la remissione completa, ovvero la scomparsa dei blasti dall'organismo. La remissione completa determina l'assenza di tutti i sintomi attribuibili alla malattia, con un normale quadro ematologico periferico, con assenza di blasti oppure inferiore al 5%. I farmaci utilizzati sono prednisone per 7 giorni, più vincristina, L-asparaginasi e methotrexate per 30 giorni.
La fase di consolidamento e reinduzione consiste nel continuare la citoriduzione e minimizzare lo sviluppo di cellule resistenti. I farmaci utilizzati sono methotrexate, citosina-arabinoside, 6-mercaptopurina, L-asparaginasi, ecc.
Infine, la fase di mantenimento agisce sulla malattia residua minima e si effettua con farmaci quali 6-mercaptopurina con methotrexate fino al completamento dei 2 anni di trattamento per tutte le fasce di rischio.
Molto importante è anche la profilassi delle complicanze del SNC. Infatti le localizzazioni neurologiche vanno in remissione completa in contemporanea alla remissione emato-midollare in oltre il 60% dei casi. Altrimenti, è possibile utilizzare chemioterapia ad alte dosi, rachicentesi con farmaci citostatici e radioterapia solo in pazienti ad alto rischio. In questo modo si ha una riduzione delle complicanze neurologiche dal 60% al 5-10% dei casi.
Leucemia mieloide acuta
La leucemia mieloide acuta è una proliferazione neoplastica delle linee mieloidi o granulocitica. La LMA rappresenta circa il 25% delle leucemie acute del bambino. Non esiste un picco per età. L'esordio è progressivo o rapido, segnato da infezione grave, una sindrome emorragica o una CID.
La classificazione si effettua tramite esame morfologico delle cellule neoplastiche. La classificazione è riportata nella tabella sottostante.
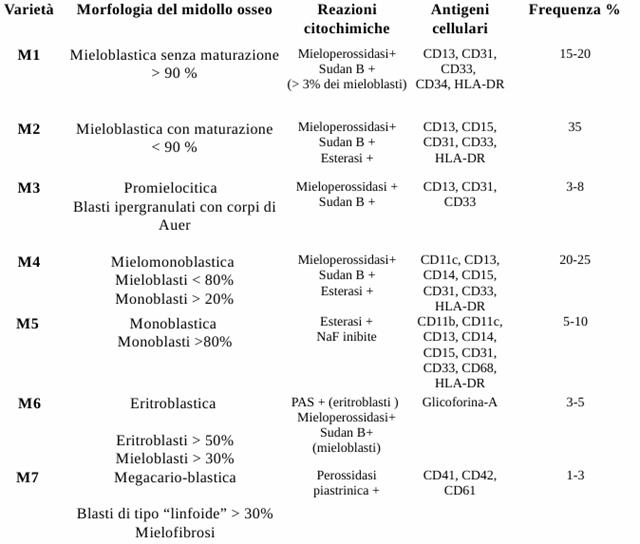
Figura : Classificazione della leucemia mieloide acuta.
Per quanto riguarda la terapia, la chemioterapia permette di ottenere l'induzione e il consolidamento. In tutte le forme i farmaci adoperati sono citosina arabinoside, antracicline ed anticorpi monoclonali. Mentre nella promielocitica (M3) si utilizzano l'acido retinoico e il triossido di arsenico. Il trapianto di cellule staminali emolinfopoietiche è consigliato nei soggetti ad alto rischio di recidiva. Nella terapia della leucemia mieloide acuta non è in genere indicata una terapia di mantenimento.
Leucemia mieloide cronica
La leucemia mieloide cronica è una proliferazione clonale di tutte le linee mieloidi con iperplasia nieloide del midollo ed ematopoiesi extramidollare. Rappresenta meno del 5% delle leucemie infantili.
![]()
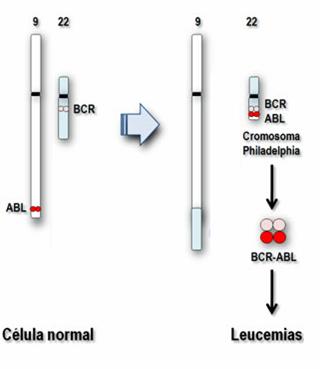 La genesi è dovuta alla presenza del cromosoma Phlidelphia
(Ph). Il cromosoma Ph deriva dalla traslocazione reciproca del braccio lungo
del cromosoma 22 sul braccio corto del cromosoma 9, detta anche
t(9;22)(q34;q11). In questo modo si forma un gene di fusione, bcr/abl, la cui
iperespressione determina la trasformazione maligna delle cellule mieloidi.
La genesi è dovuta alla presenza del cromosoma Phlidelphia
(Ph). Il cromosoma Ph deriva dalla traslocazione reciproca del braccio lungo
del cromosoma 22 sul braccio corto del cromosoma 9, detta anche
t(9;22)(q34;q11). In questo modo si forma un gene di fusione, bcr/abl, la cui
iperespressione determina la trasformazione maligna delle cellule mieloidi.
La storia naturale della malattia si articola in più fasi. Abbiamo la fase cronica, di durata di circa 3 anni, caratterizzata clinicamente da febbricola, pallore, epatosplenomegalia, dolori ossei, priapismo legato all'iperleucocitosi. Dal punto laboratoristico si osserva anemia normocromica e normocitica, leucositosi e piastrinosi. All'aspirato midollare si osserva ipercellularità e allo striscio periferico sono presenti tutti gli stadi di maturazione della linea mieloide. Alla fase cronica può seguire la fase accelerata, caratterizzata da un aggravamento della sintomatologia. Un altro scenario è dato dall'evoluzione in fase blastica, con perdita della capacità di differenziazione del clone che si traduce in leucemia mieloide acuta.
I LINFOMI
I linfomi sono neoplasie maligne, caratterizzate dalla proliferazione non controllata di cellule costituenti il tessuto linfatico, quali i linfociti, gli istiociti e i loro precursori. In realtà, con il termine di "linfoma" indichiamo un ampio gruppo di neoplasie, che possiamo suddividere in due classi:
il linfoma di Hodgkin (LH), che costituisce un'entità a sé stante, con precise caratteristiche anatomopatologiche e cliniche;
i linfomi non Hodgkin (LNH), un gruppo che presenta la caratteristica di non condividere quelle peculiarità del LH.
Linfoma di Hodgkin o LH
Per quanto riguarda il LH, rappresenta il 30% dei linfomi, con un caratteristico andamento dell'incidenza bimodale, avente il primo picco tra i 15-35 anni e un secondo picco dopo i 50 anni. Il LH è più frequente nei maschi, soprattutto nei giovani adulti. Infatti più dell'80% dei casi infantili riguardano bambini maschi. Come per ogni neoplasia, anche per il LH si sono ricercate associazioni con determinati fattori predisponenti e si è potuto notare il ruolo svolto dall'HBV. La presenza dell'HBV determina un aumento del rischio compreso tra 2-3 volte rispetto al soggetto normale.
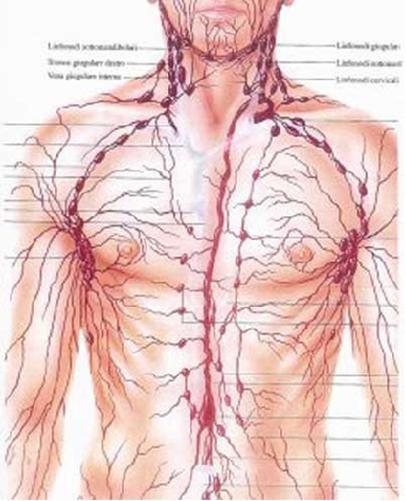 Il LH origina, nella maggioranza dei casi, in un
linfonodo o in più linfonodi della stessa stazione linfatica e si propaga
successivamente attraverso la via linfatica verso le stazioni più vicine, fino
a determinare l'invasione di tutto il sistema linfatico. Solo tardivamente il
tessuto neoplastico può invadere, attraverso la via ematica, gli organi
viscerali quali fegato, osso, polmone e cute. Le stazioni linfatiche ad essere
primariamente colpite sono quelle centrali, quindi mediastino e addome. Infatti,
non è raro riscontrare che, la sintomatologia clinica del LH è dovuta
all'effetto massa che il pacchetto linfonodale patologico determina a livello
di strutture residenti nel mediastino (esofago, trachea, cuore, ecc).
Il LH origina, nella maggioranza dei casi, in un
linfonodo o in più linfonodi della stessa stazione linfatica e si propaga
successivamente attraverso la via linfatica verso le stazioni più vicine, fino
a determinare l'invasione di tutto il sistema linfatico. Solo tardivamente il
tessuto neoplastico può invadere, attraverso la via ematica, gli organi
viscerali quali fegato, osso, polmone e cute. Le stazioni linfatiche ad essere
primariamente colpite sono quelle centrali, quindi mediastino e addome. Infatti,
non è raro riscontrare che, la sintomatologia clinica del LH è dovuta
all'effetto massa che il pacchetto linfonodale patologico determina a livello
di strutture residenti nel mediastino (esofago, trachea, cuore, ecc).
![]() Caratteristica
istologica peculiare è la presenza di una cellula gigante, detta di
Reed-Sternberg. Tale cellula, insieme anche ad altre, rappresenta l'elemento
diagnostico di LH in alcune sue forme. Si tratta di una cellula ampia, con
nucleoli che reagiscono intensamente per le colorazioni per l'RNA ed i nuclei
sono contenuti in un ampio citoplasma.
Caratteristica
istologica peculiare è la presenza di una cellula gigante, detta di
Reed-Sternberg. Tale cellula, insieme anche ad altre, rappresenta l'elemento
diagnostico di LH in alcune sue forme. Si tratta di una cellula ampia, con
nucleoli che reagiscono intensamente per le colorazioni per l'RNA ed i nuclei
sono contenuti in un ampio citoplasma.
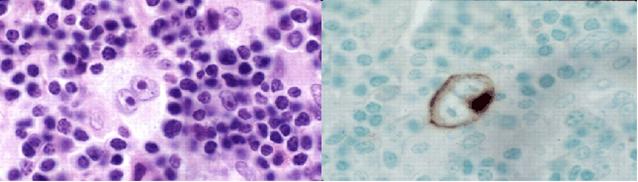
Figura : Cellula di Reed-sternberg.
Tuttavia, il riscontro bioptico della cellula RS da solo non è sufficiente per porre diagnosi di LH. Infatti, la cellula può essere presente anche in altre condizioni, quali:
mononucleosi infettiva;
carcinomi solidi;
micosi fungoide;
in alcuni LNH.
Esistono 4 varianti isto-morfologiche del LH in base alle caratteristiche delle cellule presenti:
sclerosi nodulare: rappresenta il 40-80% dei casi e presenta una prognosi eccellente;
predominanza linfocitaria: 2-10% con prognosi eccellente;
cellularità mista: 20-40%, malattia diffusa con sintomi sistemici;
deplezione linfocitaria: 2-15%, malattia disseminata ed aggressiva.
Passiamo alla clinica. In base alla presenza o meno di sintomi sistemici, tutti i linfomi vengono detti di tipo A se asintomatici, di tipo B se vi sono sintomi. I principali sintomi sistemici sono dati da febbre, sudorazione notturna e calo ponderale. Tale triade classica dei linfomi è presente in oltre il 10% dei soggetti. La presenza dei sintomi è utile ai fini della stadiazione. In circa l'80% dei casi si riscontra una tumefazione linfonodale, coinvolgendo:
linfonodi cervicali nel 70% dei casi;
linfonodi ascellari nel 10-20% dei casi;
linfonodi inguinali nel 5-10% dei casi;
stazioni linfonodali profonde, come mediastiniche, lomboaortiche, iliache, ilo epatico e splenico, in circa il 30-50% dei casi.
La milza è interessata in circa il 20% dei casi, mentre un interessamento extra-nodale si verifica nel 10%, colpendo soprattutto osso, polmone e fegato. La milza non rientra nelle sedi extranodali, in quanto è un organo linfatico.
La diagnosi si effettua con:
anamnesi: presenza o assenza dei sintomi;
E.O completo, con valutazione delle stazioni linfonodali;
laboratorio, con richiesta di esami ematochimici completi e VES.
Le principali tecniche di imaging che si possono utilizzare sono:
Rx del torace;
ecografia con power doppler, che mette in evidenza un pattern di vascolarizzazione di tipo anarchico, permettendo così la distinzione tra linfonodi maligni e benigni;
TC con mdc al collo, torace, addome e polmone;
PET/TC total body;
RMN e scintigrafia ossea.
Qualora vi sia il forte sospetto di linfoma oppure si noti la presenza di linfonodi ingrossati, si effettua una biopsia linfonodale con o meno aspirato e biopsia midollare.
Con la sola palpazione, sarà possibile effettuare la diagnosi differenziale rispetto ad altri quadri morbosi che possono determinare linfoadenomegalia. Generalmente le infezioni batteriche o virali hanno linfonodi di consistenza morbida, mobili, dolenti e assenza di fusione in pacchetti, al contrario di un linfoma o di una metastasi da tumore solido, in cui il linfonodo si presenta con una consistenza lignea, è fisso, non dolente e con fusione in pacchetti.
Una volta fatta la diagnosi, si passa alla stadiazione, in base ai criteri di Ann Arbor. In base a questa classificazione, vi sono 4 stadi. Se vi è una perdita di peso inspiegata superiore al 10% del peso abituale nei 6 mesi precedenti e/o febbre superiore ai 38°C e sudorazione notturna il paziente è sintomatico, e si aggiunge la lettera B allo stadio. Se, invece, la triade sintomatologica è assente, si aggiunge allo stadio del paziente la lettera A. Passiamo ora a descrivere i singoli stadi:
stadio I: coinvolgimento di un singolo linfonodo/stazione linfonodale (I) o di singolo sito extralinfonodale (IE);
stadio II: coinvolgimento di più stazioni linfonodali dallo stesso lato del diaframma (II) o con interessamento limitato di organi o tessuti extralinfonodali contigui (IIE);
stadio III: coinvolgimento di stazioni linfonodali su entrambi i lati del diaframma (III) che possono includere la milza (IIIS) e/o alcuni organi extralinfonodali contigui (IIIE);
stadio IV: diffusa malattia extralinfonodale (fegato, midollo osseo, polmone e cute).
Numerosi studi hanno individuato una serie di fattori in grado di predire la prognosi dei soggetti affetti da LH. I principali fattori prognostici sono:
stadio: precoce (I e II) è un fattore prognostico positivo, stadi avanzati (III e IV) sono fattori prognostici negativi;
una VES superiore a 50 è un fattore prognostico negativo;
l'interessamento di più di 4 stazioni linfonodali non contigue è un fattore prognostico negativo;
una massa linfonodale mediastinica con diametro lungitudinale massimo superiore ai 10 cm (Bulky syndrome) è un fattore prognostico negativo;
sottotipi a predominanza linfocitaria e a sclerosi nodulare rappresentano fattori prognostici positivi, al contrario delle varianti a cellularità mista e a deplezione linfocitaria.
In merito alla terapia, attualmente il LH deve essere considerato una malattia potenzialmente guaribile. La radioterapia si è dimostrata efficace in più dell'80% dei pazienti con linfoma localizzato, stadio I e II. La chemioterapia con ABVD in associazione con la radioterapia viene somministrata ai pazienti con malattia disseminata, stadi III e IV, ottendo una percentuale di successo di oltre il 50%. Il protocollo ABVD è composto da adriamicina, bleomicina, vinblastina e dacarbazina.
Linfomi non Hodgkin o LNH
Da un punto di vista epidemiologico, rappresentano circa il 2% di tutti i tumori e sono le neoplasie più comuni nei pazienti tra 20 e 40 anni. In età pediatrica sono più frequenti tra i 10-19 anni. La mortalità è stabile nonostante l'aumento dell'incidenza. Questo fenomeno è molto probabilmente dovuto ai miglioramenti dei regimi terapeutici. I LNH sono più frequenti nei maschi, con un rapporto M/F di 1,5. I principali fattori di rischio sono:
infezioni, soprattutto virali quali EBV, HTLV, HCV;
immunodepressione;
autoimmunità;
malattie genetiche.
Come i LH, i LNH possono avere origine in un linfonodo o in più linfonodi della stessa stazione linfatica e propagarsi attraverso la via linfatica e la via ematica. Ma, nel 30% dei casi, un LNH esordisce in sedi extralinfonodali quali apparato GI, cute, ossa, polmone e pleura. Il midollo osseo risulta essere la sede iniziale di malattia con una frequenza che va dal 10-40% dei casi.
La maggioranza del LNH sono di origine B (90%) e la classificazione WHO (1997) li divide in:
linfomi a basso grado di malignità;
linfomi a grado intermedio di malignità;
linfomi a grado elevato di malignità.
Solo il 20% dei casi presenta all'esordio i sintomi B che sono in ogni caso meno frequenti rispetto al LH. Più dei 2/3 presentano linfoadenopatie periferiche persistenti e non dolenti. Frequentemente sono interessati i linfonodi laterocervicali, sovraclaveari ed inguinali, a differenza dei LH, nel 30% dei casi la malattia esordisce in sede extra-nodale. Infatti, i LNH si presentano sin dall'esordio in fase più avanzata e quindi con una più diffusa compromissione linfonodale ed extra-nodale rispetto al LH.
Per la diagnosi e stadiazione vale quanto detto per il LH e la stadiazione di Ann Arbor è attualmente utilizzata anche per i LNH.
Infine, anche la terapia dei LNH si basa sull'utilizzo combinato della radioterapia e della chemioterapia, a seconda dello stadio della malattia:
nei linfomi a basso grado si utilizza la radioterapia loco-regionale;
nei linfomi a grado intermedio, radioterapia loco-regionale con polichemioterapia, schema R-CHOP;
nei linfomi ad alto grado si utilizza esclusivamente la polichemioterapia R-CHOP.
Lo schema R-CHOP prevede la somministazione di rituximab, ciclofosfamide, vincristina, antracicline e prednisone.
IMMUNODEFICIENZE CONGENITE
Si definiscono Sindromi da Immunodeficienza quelle malattie caratterizzate da alterazioni di uno o più dei 4 sistemi responsabili della difesa di un individuo da agenti estranei. Vengono classificate sulla base del tipo di difetto.
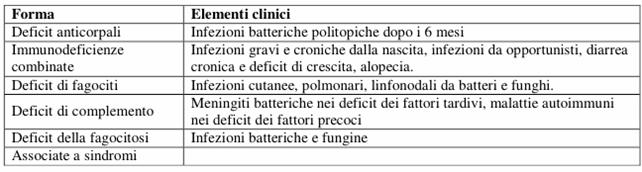
Figura : Classificazione delle immunodeficienze.
Attualmente si conoscono oltre 70 forme; ad eccezione del deficit di IgA (incidenza 1:600) sono malattie molto rare, con incidenza intorno a 1:100.000. Il bambino va incontro a infezioni gravi e ricorrenti, della categoria normalmente contrastata dal sistema di cui lui è carente; nei casi di deficit anticorpali il quadro si manifesta più tardivamente, intorno ai sei mesi di vita, perché fino a quel momento il lattante è tutelato dagli Ac di origine materna; negli altri casi si manifesta precocemente.
Nel 5% dei soggetti in età pediatrica sono riscontrabili sintomi di sospetto di una ID. Nello 0,6% dei nati vivi si riscontra una ID. Si accetta come numero normale di infezioni fino a 6 episodi/anno. Al di sopra di tale valore si parla di infezioni recidivanti. È ormai nota l'importanza dei fattori ambientali, quali l'aria molto secca e conviventi fumatori.
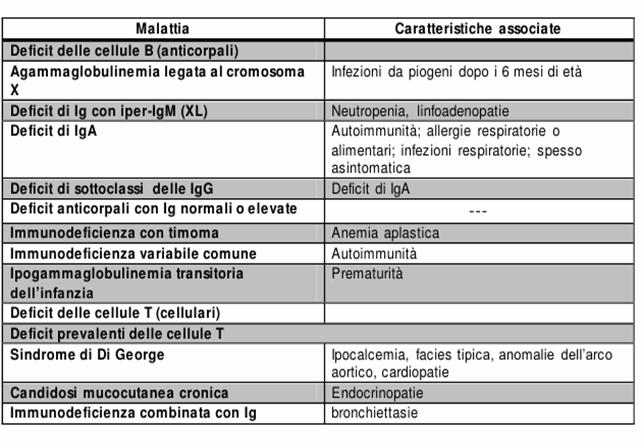
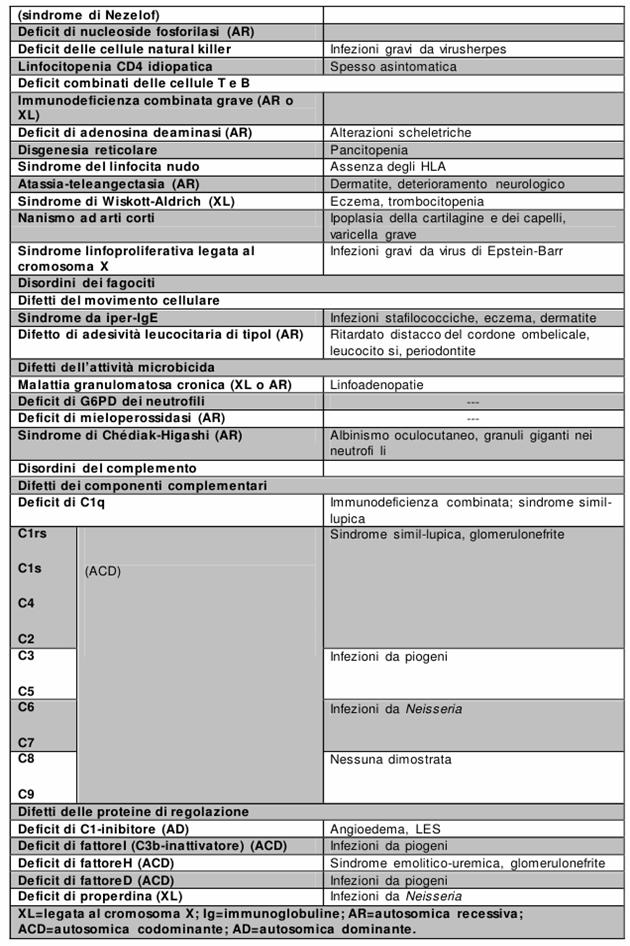
Figura : Principali caratteristiche delle ID.
Quando sospettare una ID
Anamnesi familiare
Genitori consanguinei;
Fratelli o sorelle morti precocemente;
Feti morti o aborti spontanei;
Parenti con altre malattie da Immunodeficienza.
Anamnesi personale
Infezioni molto frequenti;
Infezioni molto gravi (articolazioni, intestino, app. respiratorio);
Infezioni recidivanti con lo stesso germe;
Infezioni con germi particolari (Pneumocistis Carinii, Aspergillus, Candida, Saprofiti).
Dati clinici
Ritardo staturo-ponderale (S. da malassorbimento, diarrea intratattabile);
Ascessi anche freddi;
Mughetto;
Ipoplasia di tonsille e linfonodi (dopo i 6 mesi);
Sintomi simili al LES o alle vasculiti;
Alcune malformazioni.
Dati di laboratorio
Leucopenia;
Linfopenia;
VES bassa nonostante un'infezione in corso.
Reperti radiografici
Ipoplasia adenoidea;
Ipoplasia timica;
Usure costali;
Bronchiectasie.
Esame obiettivo
Valutazione delle condizioni generali: distrofia;
Esame del tessuto linfatico: ipo/atrofia di linfonodi e tonsille;
Obiettività delle vie respiratorie: segni di infezione cronica, perforazione timpanica, segni di broncopolmonite o segni di bronchiectasie.
Criteri orientativi per il tipo di difetto immunologico
Suggestivi di un difetto dei linfociti T
Decorso particolarmente grave di un'infezione virale usualmente lieve;
Candidosi localizzata su cute e mucose;
Linfopenia;
Diarrea intrattabile.
Suggestivi di un difetto dei linfociti B
Infezioni batteriche ricorrenti;
Sepsi o meningite;
Diarrea protratta o intrattabile.
Suggestivi di un difetto dei fagociti: Infezioni prevalentemente cutanee.

Deficit dei fagociti
ascessi cutanei;
linfonodi suppurati;
infezioni cutanee necrotizzanti;
candidosi mucocutanea cronica.
![]()
 Deficit del complemento
Deficit del complemento
infezioni da Neisseria;
LES;
infezioni da piogeni;
edema del tessuto sottocutaneo dell'apparato GI e respiratorio.
Meccanismi che determinano una condizione di anergia virus-indotta
Inappropriata apoptosi dei linfociti;
Attivazione costitutiva dei linfociti;
Effetto citopatico diretto del virus sui linfociti;
Effetto anergizzante mediato dalle cellule accessorie della risposta immune.
Quali accertamenti richiedere nel sospetto di un difetto cellulare
I livello:
Linfociti circolanti;
Multiskin test: utile nel valutare la capacità del paziente nel montare una risposta immune; si attua inoculando attraverso la cute un allergene, la risposta sarà positiva se dopo 48-72 ore sarà possibile evidenziare la presenza di un pomfo;
Rx torace.
II Livello:
Conta linfociti T, helper e suppressor;
Determinazione enzimatica;
Tests funzionali in vitro (test clonogenici).
Cosa non si deve mai fare ad un malato in cui si sospetta un' ID
Trasfusioni di sangue;
Vaccinazioni con virus vivi (Poliomielite orale, Morbillo, Rosolia).
Difetti dell'immunità specifica

Agammaglobulinemia
L'agammaglobulinemia è una patologia ereditaria di cui riconosciamo due forme:
Agammaglobulinemia X-linked o malattia di Bruton: il gene anomalo mappa su q22 sul braccio lungo del cromosoma X e codifica per la proteina ad attività tirosinchinasica Btk (tirosinchinasi di Bruton) delle cellule B;
Agammaglobulinemia autosomica recessiva, di cui non si conosce ancora il difetto associato.
Diagnosi
Linfociti B: < 1%;
IgG sieriche: < 100% mg/dl;
IgA ed IgM: assenti;
Isoagglutinine: assenti;
Titoli anticorpali: assenti
Nel fare la diagnosi, in questa come in tutte le altre immunodeficienze, sarà importante ricordare che, essendo il sistema immunitario del bambino in via di sviluppo, i livelli delle cellule circolanti andranno confrontati con specifici range di valori adatti all'età del paziente.


Immunodeficienza Combinata Grave (SCID)
È la forma più frequente (50-60 %): è causata da mutazioni nel gene che codifica per IL-2-R-γ catena (che è in comune con il recettore di numerose citochine, IL-4, IL-7, IL-9, IL-15, indispensabili per la maturazione dei T linfociti). Si associa a deficit di ADA (accumulo di adenosina tossica per i linfociti immaturi).
Esordio: primi mesi di vita.
Problemi clinici predominanti
Eritrodermia;
Epatosplenomegalia;
Adenomegalia.
Eventi temibili:
Anasarca da protido-dispersione;
Sepsi cutanee.
Alterazioni immunologiche:
Difetto combinato T/B (T+/B-);
Linfocitosi con T attivati;
Uso limitato di specificità TCR;
Intensa eosinofilia;
Cloni T autorettivi.
Molto frequentemente è asintomatica.
Trattamento delle SCID: Nei pazienti in attesa del trapianto:
Terapia sostitutiva con Ig endovena;
Iperalimentazione per via parentale (a bambini presentano un metabolismo molto attivo, con conseguente eccesso di consumo energetico);
Co-trimossazolo per la profilassi dell'infezione da Pneumocistis;
Ketoconazolo per la candidasi;
Trapianto:
Midollo da fratello HLA-identico (matched sibling donor);
Midollo da familiare aploidentico depleto di cellule T (unmatched);
Midollo da donatore non parente depleto di cellule T (MUD).
Per il deficit di ADA oggi si sta tentando la terapia genica; alcuni laboratori stanno lavorando in questo senso.
Sindrome Di OMEN
È una forma di SCID autosomica recessiva in cui i neonati mancano sia dei linfociti B che dei linfociti T e sono presenti in circolo unicamente cellule NK (B-, T-, NK+). È causata dalla mutazione dei geni che attivano la ricombinasi RAG1 e RAG2; queste mutazioni determinano un'incapacità di formare i recettori degli antigeni attraverso la ricombinazione genetica.
Sindome Di Wiskott-Aldrich
Recessiva, legata al cromosoma X;
Il gene anomalo codifica per la "proteina della sindrome di Wiskott-Aldrich (WASP), che causa disfunzioni del citoscheletro;
Disfunzione delle cellule B e T;
Sintomi associati:
Trombocitopenia e petecche;
Eczema;
Epatosplenomegalia;
Anemia emolitica autoimmune;
Alta incidenza di linfoma - CNS.
Trattamento: cellule staminali o trapianto di midollo osseo.
Prognosi: l'aspettativa di vita è, in media, 11 anni.
Malattie da difetto del sistema di trasduzione
Atassia - teleangectasia
Patologia autosomica recessiva (11q22-23), associata a "mutazione da atassia teleangectasica" (ATM), con formazione di un prodotto molto simile PI3chinasi.
Ereditarietà: AR.
Esordio sintomi: 2-4 anni.
Alterazioni immunologiche
Difetto T cellulare;
Difetto di IgA e di sottoclassi di IgG.
Principali sintomi
Atassia (cerebellare);
Teleangectasie (cutanee e oculari);
Infezioni, Tumori, Diabete;
Ritardo di accrescimento.
Peculiarità: sviluppo linfomi e/o altre neoplasie (fino al 30-40% dei casi).
Alterazioni citogeniche: elevato numero di traslocazioni ed inversioni in punti di rottura preferenziale (per fragilità cromosomica).
Prognosi: infausta a distanza di tempo.
Diagnosi
Quadro clinico chiaro;
Aumento α-fetoproteina;
Fragilità cromosomica.
Sindrome di De George
Insufficiente sviluppo del III e IV arco branchiale con ipoplasia di timo e paratiroidi, anomalie cardiache (tronco comune) e facciali (micrognazia, ipertelorismo, inserzione bassa delle orecchie, frenulo nasale corto).
Difetto genetico autosomico dominante: delezione interstiziale 22q11.
Gravità clinica varia: da grave a parziale linfopenia con moderata immunodeficienza.
|
| Appunti su: leucemia pediatrica, classificazione delle leucemie in etC3A0 pediatrica, esordio leucemia linfoblastica acuta, linea linfoide e mieloide, |
|
| Appunti Nutrizione |  |
| Tesine Bellezza |  |
| Lezioni Bambini |  |