|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 4106 | Gradito: |
Leggi anche appunti:Fiosiologia respiratorioFIOSIOLOGIA RESPIRATORIO 300 milioni di alveoli superficie di scambio(area TetanoTetano Il tetano è una tossinfezione dovuta all'esotossina di Clostridium Adenoma Pleomorfo (un tempo definito tumore misto)Adenoma Pleomorfo (un tempo definito tumore misto) E' la neoplasia |
 |
 |
EPATOLOGIA PEDIATRICA
Ipertransaminasemia
Gli indici laboratoristici della funzionalità epatica sono:
Transaminasi o aminotransferasi (indicatori di lesione epatocellulare):: ALT (alanina-aminotransferasi)/GPT (glutammico-piruvico transferasi), maggiormente epatospecifica; AST (aspartato-aminotranferasi)/GOT (glutammico-ossalacetico transferasi), meno epatospecifica, in quanto presente anche in muscolo scheletrico, cuore, rene, cervello, globuli rossi;
Fosfatasi alcalina (PA), essendo presente in molti tessuti, per identificarne la provenienza è necessario effettuare il dosaggio degli isoenzimi PA: Biliare (PA1), aumenta nella patologia biliare; osseo/epatico (PA2); intestinale/placentare (PA3);
γ-GT (γ-glutamil-transpeptidasi), enzima microsomiale, presente negli epatociti e nell'epitelio biliare è al contempo indicatore di: danno epatocellulare (se associato ad aumento transaminasi); ostruzione biliare (se associato ad aumento PA);
LDH, enzima citoplasmatico presente in vari tessuti (cuore, muscolo scheletrico, GR) tra cui il fegato, indice di danno epatocellulare;
Indicatori di sintesi epatica (ed indirettamente di danno epatocitario), livelli sierici di albumina e proteine plasmatiche e Quadro Proteico Elettroforetico (QPE), PT o INR.
Una ipertransaminasemia può riconoscere una delle seguenti cause:
cause infettive (HAV, HBV, HCV, EBV, CMV );
cause autoimmuni (AIH/PSC);
cause metaboliche/genetiche (es. m. di Wilson, deficit α1-antitripsina, etc.);
cause nutrizionali/intestinali (celiachia, obesità, MICI);
cause tossiche;
malattie muscolari.
Circa i 2/3 delle ipertransaminasemie si risolvono entro 6 mesi, tali ipertransaminasemie sono secondarie a: epatiti da virus epatotropi minori, epatotossicità da farmaci, epatopatia da causa non nota. In 1/3 dei casi, al contrario, i livelli ematici di transaminasi persistono elevati per più di 6 mesi: forme secondarie ad obesità, ad epatiti autoimmuni, ad epatopatie genetiche (glicogenosi, miopatia, deficit di α1-antitripsina, Morbo di Wilson, IEF) e forme criptogenetiche (idiopatiche).
La corretta diagnosi prevede i seguenti step:
Anamnesi;
Esame obiettivo;
Laboratorio: I e II livello;
Score diagnostici;
Cause frequenti;
Cause meno frequenti ed emergenti..
La corretta diagnosi in un bambino con ipertransaminasemia è importante per diversi aspetti:
Ci permette di praticare un precoce e corretto intervento terapeutico. Ciò è essenziale soprattutto nelle patologie silenti, rapidamente progressive, come le epatiti autoimmuni o il morbo di Wilson, che, se non curate rispettivamente con corticosteroidi e con D-penicillamina, evolvono in cirrosi epatica e nel morbo di Wilson avanzato (encefalopatia);
Possibilità di instaurare misure di prevenzione. Ad es., in seguito ad un'infezione la malattia può essere trasmessa per via orizzontale o verticale mediante il canale del parto (es. una paziente che non sa di essere portatrice di HBV può contagiare il figlio al momento del parto); in seguito ad adozioni internazionali sono molto frequenti casi di trasmissione da bambini, con ipertransaminasemia da epatopatia virale di tipo B o C ai genitori adottivi;
Infine la diagnosi è importante per le malattie genetiche ai fini di indirizzare i pazienti e le famiglie al counseling genetico.
Anamnesi: L'anamnesi è fondamentale per formulare l'ipotesi diagnostica. In base alle suddette possibili eziologie, l'anamnesi dovrà investigare i seguenti aspetti, per ciascuna possibile causa. Nell'anamnesi familiare valutiamo anche se i genitori hanno patologie epatiche.
Esame obiettivo: L'esame obiettivo è fondamentale e deve essere generale e non solo epatico. L'esame obiettivo del fegato deve valutare le dimensioni (eventuale epatomegalia), la consistenza (parenchimatosa o meno, ad es. lignea), il contorno (regolare o meno, ad esempio nodulare), la dolorabilità, la presenza di masse epatiche. E'importante, inoltre, documentare la presenza di eventuali segni indicativi di ipertensione portale (epatopatia cronica/cirrosi) come: splenomegalia, ascite, shunt porto-cavali (causa, ad es. di sanguinamento di varici esofago-gastriche), spider naevi ed eritema palmare.
La sindrome epato-renale è definita come un'insufficienza renale funzionale in pazienti con epatopatia terminale. La sindrome epato-polmonare è caratterizzata da una tipica triade: ipossiemia, dilatazione vascolare intrapolmonare ed epatopatia. Questa sindrome comporta uno shunt di sangue intrapolmonare destro-sinistro con conseguente desaturazione sistemica.
L'encefalopatia epatica può coinvolgere qualsiasi funzione neurologica. Il suo esordio può essere eclatante o subdolo. La sua comparsa dipende dalla presenza di shunt porto-sistemici, da alterazioni della BEE e da interazioni con metaboliti tossici con il SNC.
Una colestasi, infine, può manifestarsi all'esame obiettivo con ittero, xantomi e xantelasmi, lesioni cutanee da grattamento.
Da non sottovalutare l'associazione anche di un lieve ittero con ipocromia delle feci ed ipercromia delle urine (segni indicativi di epatopatia).
Come già detto l'esame obiettivo deve essere generale e non solo epatico, di fatti i pazienti con patologie genetico-metaboliche si caratterizzano per specifici dimorfismi (basti pensare al paziente con glicogenosi, con la caratteristica faccia a bambola, che si accompagna a steatosi ed epatosplenomegalia). I pazienti affetti da NAFLD/NASH sono obesi (ed all'ecografia presentano fegato brillante, che in seguito a dieta ed attività fisica ritorna isoecogeno, per la reversibilità della steatosi). L'obesità è attualmente molto diffusa, dunque bisogna non associare l'obesità alla sola NAFLD/NASH, in quanto possono essere presenti casi in cui il bambino presenta obesità associata a: morbo di Wilson, celiachia (es. un bambino con una celiachia patchy), epatite autoimmune, ecc..
I pazienti distrofici (i troppo magri) o di bassa statura potrebbero essere affetti da celiachia. La bassa statura può anche essere la spia di una Sindrome di Turner, nella quale non sono rare le complicanze epatiche (motivo per il quale, nel management di queste pazienti dovrebbe essere presa in considerazione anche la valutazione della funzione epatica). L'epatite autoimmune può associarsi ad altre manifestazioni autoimmuni, come la vitiligine.
Il Wilson si associa ad alcuni segni clinici caratteristici quali:
anello di Keyser-Fleisher dovuto alla precipitazione di sali di rame in corrispondenza della membrana di Descemet della cornea; appare all'esame con lampada a fessura come un anello marrone-verdastro alla periferia della cornea;
segni di interessamento neurologico tremori, distonie, disartria, . . .;
lunula cerulea (lunule ungueali bluastre).
La cataratta si presenta nei pazienti in seguito ad infezioni connatali o in presenza di galattosemia. L'ippocratismo digitale può essere presente in alcune patologie: CF, Cirrosi, MICI, Colestasi croniche.
La linfoadenomegalia è presente in pazienti con infezione da CMV, EBV, Toxoplasma.
Per
un paziente in cui sospettiamo una miopatia dobbiamo valutare la pseudoipertrofia
dei polpacci accompagnata ad ipotonia (dovuta a sostituzione del tessuto
muscolare con tessuto fibro-adiposo) ed ![]()
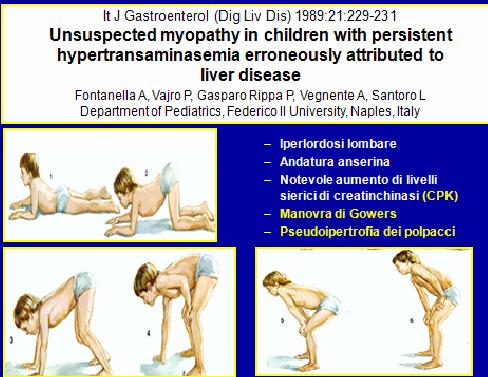 effettuare la manovra di Gowers: valuta il
numero di movimenti che il paziente esegue per svolgere una determinata azione;
in genere, si chiede al paziente di alzarsi dal pavimento e si considera il
segno positivo se egli si alza con il tronco flesso in avanti, facendo forza
con le mani sulle ginocchia.
effettuare la manovra di Gowers: valuta il
numero di movimenti che il paziente esegue per svolgere una determinata azione;
in genere, si chiede al paziente di alzarsi dal pavimento e si considera il
segno positivo se egli si alza con il tronco flesso in avanti, facendo forza
con le mani sulle ginocchia.
Indagini di laboratorio: Test di I livello:
γ-GT: per valutare una patologia delle vie biliari (colestasi, colangite sclerosante);
QPE: Il pediatra richiede il quadro siero-proteico: per valutare se il pz. presenta un'iper-γ-globulinemia, indice di epatite autoimmune, per valutare la presenza di ipo-albuminemia, indice di epatopatia avanzata con ridotta protidosintesi (cirrosi), o per valutare l'assenza del picco delle α-1-globuline indice della presenza del deficit di α-1-anti-tripsina;
IgG, IgA ed IgM: si prescrivono per valutare la presenza di un eventuale deficit di IgA, che frequentemente si associa a celiachia;
HCV-Ab: per l'epatite da HCV; ed HBsAg ed HBcAb per l'HBV. Di solito il titolo di HBsAb è maggiore di 50-100 U/L in quanto si effettua la vaccinazione contro HVB;
Anticorpi anti virus epatotropi minori (Ab Anti CMV/EBV);
Anticorpi anti-endomisio (EMA), anti-transglutaminasi tissutale (tTGA) ed anti-gliadina (AGA) per la diagnosi di malattia celiaca. Dal 1991 è stato stabilito che nei bambini di età inferiore ai 2 anni bisogna valutare gli AGA, in quanto non vengono formati ancora gli EMA e i t-TGA, che sono quindi meno attendibili. Inoltre gli EMA ed i t-TGA sono IgA e risultano negativi in pz. con deficit di IgA. Quindi gli AGA vanno valutati in pazienti di età < 2 anni e in quelli in cui si sospetta un deficit di IgA;
Sideremia/transferrina: per l'emocromatosi, nella quale la sideremia è aumentata e la transferrina libera è diminuita (si riduce la T.I.B.C.). Una transferrina libera elevata ci permette, dunque, di escludere una emocromatosi. Nel bambino si definisce anemia quando l'Hb è < 10,5-11 mg/dl;
Ceruloplasmina sierica/cupremia: per la valutazione della malattia di Wilson; la ceruloplasmina sierica è diminuita e la cupremia aumentata;
CPK: per escludere la presenza di una miopatia. Si monitora il valore della CPK perché la distrofia muscolare di Duchenne e di Becker inizialmente sono asintomatiche in quanto la necrosi delle fibre muscolari non è ancora elevata. In presenza di CPK e aldolasi elevate, si sospetta una patologia muscolare e quindi l'ipertransaminasemia è di origine muscolare e non epatica;
Clini test: dosaggio delle sostanze riducenti nelle urine (galattosio, fruttosio, glucosio), per escludere galattosemia ed eventuale Intolleranza Ereditaria al Fruttosio (I.E.F.).
Test di II livello:
ANA (anticorpi-anti nucleo), ASMA (anticorpi anti-muscolo liscio), LKM1 (anticorpi anti-liver/kidney microsomes) e LC1 (anticorpi anti-citosol epatico), effettuati per escludere un'epatite autoimmune (AIH). Gli LKM sono anticorpi contro i microsomi epatici e renali. Sono anticorpi non organo-specifici. Gli LC1 sono stati individuati di recente come causa di epatite autoimmune anche in età pediatrica;
Gli ANCA (anticorpi anti-citoplasma dei neutrofili) associati alla colangio-RM per la colangite sclerosante e, in associazione ai suddetti auto-anticorpi per l'AIH, per le sindromi da overlap tra epatite autoimmune e colangite sclerosante (colangiti autoimmuni). Nella colangite tipico è l'aspetto a corona di rosario con restringimenti e dilatazioni che possono essere valutate mediante una colangio-RM;
In un bambino che presenti markers epatitici B e C negativi, è importante anche effettuare l'HBV-DNA, l'HCV-RNA e il genotipo in quanto ci sono delle forme di epatite in cui sono presenti i cosiddetti "santuari", rappresentati da linfociti. Il virus HCV può rifugiarsi all'interno dei linfociti oppure l'HBV può andare direttamente nel fegato senza che sia presente una sierologia positiva. Queste forme vengono anche chiamate criptiche. Si possono manifestare maggiormente in pazienti che sono sottoposti a terapia antiblastica;
In caso di deficit di α1-antitripsina (già sospettato per il riscontro al QPE del deficit di α-1-globuline) viene valutato: il livello sierico di α1-antitripsina con metodo immunonefelometrico; viene dosata la proteina mediante l'utilizzo di un anticorpo e mediante il metodo turbinimetrico ne viene dosata la concentrazione; il fenotipo, possiamo avere una forma omozigote mZZ o PiZZ (Protease Inhibitor ZZ) con carenza completa della proteina in circolo, oppure forme eterozigoti: mz o ms. PiZ-, PiZS, PiZI;
Indagini di II livello nella diagnostica del Wilson sono: la cupruria basale, il paziente con Wilson ha un aumento della concentrazione di rame nelle urine in condizioni basali; la cupruria dopo test di sensibilizzazione con penicillamina. La penicillamina è una resina che lega il rame e ne fa aumentare il livello urinario. Il test viene effettuato mediante la raccolta delle urine delle 24h; queste ultime devono essere raccolte in un contenitore di plastica e non di vetro (perché contiene tracce di rame). All'interno del contenitore deve essere presente un po' di acido cloridrico che evita la fermentazione delle urine. Si effettua successivamente una seconda raccolta delle urine delle 24h somministrando al paziente la penicillamina al mattino e alla sera. Si può valutare l'aumento della concentrazione urinaria di rame nella seconda raccolta delle urine; un altro test che può essere effettuato è il test molecolare che ricerca la mutazione ATP7B presente nel morbo di Wilson;
La ferritinemia per l'emocromatosi; in caso di emocromatosi sarà aumentata. Il focusing isoelettrico della transferrina viene effettuato per ricercare la transferrina carboidrato-carente e valutare, quindi, disordini congeniti della glicosilazione delle proteine, CDG. L'alterazione della glicosilazione è sistemica e si manifesta anche a livello epatico;
Il test del sudore (che valuta la concentrazione di cloro nel sudore) si effettua per valutare la fibrosi cistica;
Possiamo effettuare il test molecolare IEF in caso di sospetta intolleranza al fruttosio;
L'ammonemia, l'amminoacidemia ed equilibrio acido-base vengono valutati in caso di sospetto di disordini del ciclo dell'urea.
Indagini strumentali: L'ecografia può fornire informazioni sulle dimensioni e sul flusso ematico del fegato. Ha un ruolo importante nella valutazione della steatosi epatica (fegato iperecogeno, con spot ipoecogeni in vicinanza degli spazi portali), ma anche in caso di cirrosi o lesioni focali epatiche. Per quanto riguarda la patologia delle vie biliari, ha sostituito la colangiografia nella diagnosi di litiasi biliare e della colecisti. Può consentirci anche di individuare dilatazioni cistiche delle vie biliari. Permette inoltre di valutare alterazioni delle vie biliari persino nel neonato.
La colangio-Rm è estremamente utile nella diagnosi differenziale della patologia delle vie biliari (ad es. colangite sclerosante). In caso di neoplasia epatica la TC rappresenta la migliore indagine strumentale.
La colangiografia percutanea trans epatica con ago sottile è la metodica di scelta per lattanti e bambini piccoli; è stata utilizzata per delineare il sistema duttale biliare.
La biopsia epatica: La biopsia epatica, combinata con i dati clinici, consente di raggiungere una diagnosi eziologica, nella maggior parte dei casi.
I campioni ottenuti possono essere utilizzati per:
la valutazione istopatologica in pazienti con malattie metaboliche, colestasi neonatale, epatite cronica attiva, ecc.;
la valutazione ultrastrutturale al M.E., ad esempio, nella malattia di Wilson, la microscopia elettronica ci consente di apprezzare modificazioni ultrastrutturali mitocondriali (granulazioni o incremento della densità della matrice);
l'analisi enzimatica, in caso di sospetto di errori congeniti del metabolismo;
la ricerca di sostanze da accumulo, come il rame (tramite coloranti come la rodanina o l'orceina), il ferro, l'α1-antitripsina (che appare come granuli PAS-positivi e diastasi resistenti all'interno del RE);
effettuare analisi molecolari;
monitorare la risposta alla terapia o eventuali complicanze di un trattamento epatotossico.
La biopsia è indicata in caso di ipertransaminasemia persistente da più di 6 mesi, o ancor prima se si sospetti un Wilson o una epatite autoimmune, per le quali la terapia deve essere impostata precocemente, prima dei sei mesi, in quanto si tratta di una terapia salva vita.
Istopatologia: La biopsia è importante perché ci dà informazioni sul grading, il grado di infiammazione, e sullo staging, entità della fibrosi. È importante anche per la valutazione diagnostica, ad esempio:
l'epatite attiva è presente nella malattia celiaca: gli spazi portali presentano un infiltrato linfoplasmacellulare; quando l'infiltrato fuoriesce dallo spazio portale erode la lamina limitante ed invade il lobulo circostante, quanto maggiore è lo spill over dei linfociti e dei monociti maggiore è l'attività. Viene definita spilling necrosis;
la steatosi epatica ci orienta verso una malattia dismetabolica (NAFLD, l'intolleranza al fruttosio, morbo di Wilson). Tuttavia sono stati descritti casi di celiachia associati ad obesità con fegato steatosico;
una fibrosi concentrica periduttale (onion skin fibrosis = fibrosi a bulbo di cipolla) ci orienta verso una colangite sclerosante primitiva.
È controverso l'uso generalizzato della biopsia: nelle epatopatie correlate ad obesità e nelle epatiti croniche virali di pz. non candidati a terapie antivirali. Di fatti nei pazienti con Epatopatia correlata all'Obesità, nelle prime fasi il trattamento si basa sulle modifiche dello stile di vita (dieta ed attività fisica). Se il pz. perde 2 kg/mese possiamo assistere ad una progressiva riduzione delle transaminasi verso i v. n. La biopsia in un paziente obeso va effettuata, quindi, solo se: si sospetti un'evoluzione verso una epatopatia cronica progressiva (quindi in presenza di splenomegalia, fegato di consistenza aumentata, ecc.); se si ha il sospetto di una patologia dismetabolica (es. Morbo di Wilson).
Le Epatiti Virali possono essere trattate con interferone, lamivudina, ribavirina. In questi casi la biopsia dovrebbe essere effettuata prima del trattamento per valutare la situazione di partenza. In età pediatrica l'epatite virale B e C non sono rapidamente progressive, per cui l'atteggiamento terapeutico è di tipo attendista. L'80% dei pazienti di età pediatrica vanno incontro a risoluzione dell'epatite da HBV prima del raggiungimento dell'età adulta, ciò implica che hanno un meccanismo di clearance del virus, accelerato dal trattamento con interferone. Se i genitori del paziente si oppongono al trattamento si interviene solo in caso di viraggio del quadro clinico.
Le controindicazioni alla biopsia comprendono un aumento del PT o dell'INR, trombocitopenia, sospetto di lesioni di natura vascolare, cistica o infettiva e ascite tesa.
Epatopatia correlata ad obesità (Non Alcoholic Fatty liver disease/ Non Alcoholic Steato-Hepatitis)
Parallelamente all'epidemia di sovrappeso ed obesità, che negli ultimi 20 anni ha interessato la popolazione pediatrica di tutto il mondo, si è riscontrato un incremento della prevalenza della Non-Alcoholic Fatty Liver Disease (NAFLD), quale principale causa di epatopatia cronica nel bambino e nell'adolescente nei Paesi industrializzati. L'acronimo NAFLD identifica uno spettro di condizioni clinico-istologiche, che vanno dalla steatosi epatica, semplice accumulo di trigliceridi all'interno dell'epatocita, alla steatoepatite non alcolica (NASH), caratterizzata anche da una componente infiammatoria, fino a forme presentanti una componente fibrosa più o meno marcata ed evolventi a cirrosi ed insufficienza epatica, anche in età pediatrica. Risulta semplice, dunque, comprendere come una diagnosi tempestiva ed una adeguata terapia siano fondamentali ad ogni età, ma ancor più nel bambino/adolescente, ai fini di prevenire l'evoluzione verso una grave epatopatia nell'adulto.
Patogenesi: Nonostante la patogenesi non sia ancora completamente nota, gli studi condotti fanno supporre che il meccanismo preveda due insulti: il primo, caratterizzato dall'accumulo di grasso all'interno degli epatociti (NAFLD semplice), sarebbe capace di sensibilizzare il fegato al secondo insulto che conduce alla necro-infiammazione (NASH) e quindi alla fibrosi. Il disordine metabolico chiave nel primo step è l'alterazione del metabolismo lipidico a favore della lipogenesi rispetto alla lipolisi, che è la diretta conseguenza metabolica di una condizione di insulino-resistenza che accomuna questi pazienti. Ricordiamo, inoltre, che l'insulino-resistenza è il comune denominatore eziopatogenetico di un gruppo di condizioni patologiche, che nell'insieme costituiscono la sindrome metabolica, vale a dire: intolleranza al glucosio fino al diabete di tipo II; obesità; ipertensione; dislipidemia. Secondo alcuni, la NAFLD non sarebbe altro che la manifestazione epatica della suddetta sindrome.
Il fegato steatosico è più suscettibile, rispetto al fegato normale, all'esposizione a fattori nocivi, che comportano l'innesco di un processo infiammatorio tissutale, con:
stress ossidativo e conseguente perossidazione lipidica;
produzione e rilascio di citochine pro-infiammatorie e pro-fibrotiche.
I fattori responsabili della progressione della steatosi verso le lesioni gravi della steatoepatite e della fibrosi sono ancora poco definiti; tra essi sta assumendo importanza notevole l'alterazione dell'asse fegato-intestino (microbiota intestinale), che comporterebbe un aumento della permeabilità della mucosa intestinale, con assorbimento di endotossine ed altri prodotti batterici che andrebbero ad innescare il processo infiammatorio a livello epatico.
Istopatologia della NAFLD pediatrica: Sia nel bambino che nell'adulto la diagnosi di NAFLD non è possibile senza il ricorso alla biopsia epatica.
Nell'adulto le caratteriste istologiche che rappresentano la NASH sono ampiamente descritte: steatosi macrovescicolare, infiammazione globulare e la degenerazione balloniforme degli epatociti, spesso con pochi corpi ialini di Mallory, e talora deposito di collagene perinusoidale e perivenulare.
Gli studi effettuati su diverse coorti di bambini hanno dimostrato che alcuni aspetti istologici sono simili a quelli riscontrati nei pazienti adulti, mentre altri aspetti sono peculiari della NAFLD pediatrica. Nel bambino, infatti, troviamo come nell'adulto steatosi con balloning e/o fibrosi perisinusoidale in assenza di elementi di alterazione portale; ma troviamo anche steatosi associata ad infiammazione portale e/o fibrosi in totale assenza di fibrosi perisinusoidale o balloning.
Diagnosi di NAFLD/NASH: Le evidenze descritte, sottolineano l'esigenza di una diagnosi precoce di NAFLD in bambini ed adolescenti, in modo da prevenire il possibile sviluppo di gravi patologie epatiche degenerative sia in età pediatrica che successivamente in età adulta.
Attualmente, nella maggior parte di questi pazienti la diagnosi di NAFLD nasce dal riscontro casuale, in pazienti asintomatici, di alterazioni degli indici di funzionalità epatica (transaminasi, in particolare ALT, e gamma-GT), in associazione al rilievo ecografico di "bright liver" ovvero fegato iperecogeno, con spot ipoecogeni nelle aree periportali, aumentato in dimensioni ed a margini smussi.
Comunque, la biopsia epatica rappresenta il "gold standard" per ogni valutazione e stadiazione di NAFLD/NASH, ma, è comunque una tecnica invasiva.
Nella tabella vengono riassunte le attuali tecniche non invasive per la diagnosi di NAFLD/NASH.
|
Indici Bioumorali. |
Livelli di ALT e di gamma-GT. Questi parametri non risultano essere sempre efficaci in quanto anche in pazienti con NAFLD istologicamente accertata, il valore degli enzimi epatici può anche risultare normale. |
|
Ecografia Addominale. |
E' il test di screening più comunemente usato per la diagnosi di fegato grasso. Tuttavia la sensibilità dell'esame è limitata dall'incapacità di rilevare l'infiltrazione di grasso al di sotto del 33%, di differenziare i diversi gradi della malattia e di rilevare la presenza di NASH. |
|
Tomografia computerizzata. |
E' più sensibile e più specifica dell'ecografia, e può risultare utile nel monitorare il contenuto in grasso. Il suo limite maggiore risiede nel possibile effetto maschera prodotto dalla presenza di un sovraccarico di ferro, frequente nella NAFLD, che ne diminuisce la sensibilità. |
|
Risonanza magnetica nucleare. |
È sicuramente la tecnica d'immagine più sensibile e più specifica per la diagnosi di NAFLD. Non permette di distinguere fra semplice steatosi e steatoepatite, più o meno complicata da fibrosi, e rimane comunque una tecnica di alto costo. |
|
Tecniche non-invasive in corso di valutazione. |
Tra le nuove metodiche in fase di valutazione: i marcatori della matrice extracellulare che sono legati alla formazione di fibrosi e allo sviluppo di cirrosi epatica, come ad esempio gli "ELF MARKERS"; l'elastografia transiente (Fibroscan) che permette una buona valutazione della progressione della malattia. |
Terapia: Il caposaldo della terapia è rappresentato dalle modifiche dello stile di vita (dieta ed attività fisica), finalizzate alla perdita di peso graduale e controllata (5-10% del peso corporeo basale; 2 kg/mese). Nei pazienti non complianti, che non riescano a raggiungere il target terapeutico con le sole modifiche dello stile di vita, si rende necessario un approccio farmacologico multidisciplinare basato su farmaci che agiscono sui meccanismi eziopatogenetici, come:
metformina (insulino-sensibilizzanti);
antiossidanti;
acido ursodesossicolico UDCA (citoprotettivo).
Nuovi approcci terapeutici potranno essere rappresentati da: incretine ed inibitori del DPP-4 (aumentano la secrezione insulinca), omega-3 (DHA), probiotici, pentossifillina ed anti-TNFα, agonisti FXRs, TLRs modifiers, ecc. . .
Epatite Autoimmune (AIH)
L'epatite autoimmune è un processo infiammatorio cronico che si manifesta con ipertransaminasemia, iper-Ig e presenza di autoAb sierici associati al fegato. L'ipoalbuminemia è frequente, il PT è aumentato spesso come conseguenza del deficit di vitamina K. Sono di frequente riscontro un'anemia normocromica e normocitica, leucopenia e trombocitopenia. Il bersaglio del processo infiammatorio possono essere sia gli epatociti che l'epitelio dei dotti biliari. La gravità della manifestazione della patologia è variabile. Nella maggior parte dei casi l'esordio è insidioso.
È possibile distinguere 3 sottotipi di epatite autoimmune:
Tipo I presenza di ANA e/o ASMA, si presenta in età adulta nel 70% dei casi come epatite cronica e nel restante 30% come epatite acuta;
Tipo II presenza di anticorpi LKM e anti-LC1, si presenta in età giovanile nel 30-40% dei casi con esordio acuto e nel 60% con modalità simile al tipo I dalla quale si differenzia per la più rapida evoluzione in cirrosi e la più frequenti recidive;
Tipo III presenza di anticorpi anti SLA/LP, si presenta generalmente in giovani donne con un quadro simile al tipi I.
Criteri Diagnostici (1999)
sesso femminile;
presenza di altre malattie autoimmuni:
ipertransaminasemia prevalente rispetto alla γ-GT o ALP:
presenza di iper-γ-globulinemia;
presenza di auto Ab (ANA, ASMA, LKM1);
assenza di marcatori epatitici;
assenza di assunzione di tossici, alcol;
assenza di patologie genetiche;
presenza di HLA DR3-DR4;
istologia di epatite attiva;
trattamento cortisonico riduce i livelli di transaminasi (risposta alla terapia I.S./relapse a sospensione)
Questo score permette vari gradi di certezza diagnostica: dalla diagnosi probabile alla diagnosi certa.
Terapia: prednisone (2 mg/kg/die) + azatioprina (1-2 mg/kg/die), terapia di combinazione che consente di ridurre il dosaggio dello steroide, limitandone gli effetti collaterali. Nei pz pediatrici qualche risultato si è avuto anche con la ciclosporina A.
Malattia di Wilson
 La Malattia di Wilson (WD), anche conosciuta come
DEGENERAZIONE EPATO-LENTICOLARE, è un disordine autosomico recessivo del
metabolismo del rame, metallo essenziale, importante cofattore di molte
proteine. L'incidenza è stimata essere di 1/50.000-1/80.000. Il gene della
malattia codifica per una adenosina trifosfatasi 7B (ATP7B) espressa primariamente
dal fegato, ma anche da cervello, rene e placenta. Le mutazioni causa della malattia
conducono ad un difetto della escrezione del rame nella bile (via principale di
escrezione del metallo dal nostro organismo).
La Malattia di Wilson (WD), anche conosciuta come
DEGENERAZIONE EPATO-LENTICOLARE, è un disordine autosomico recessivo del
metabolismo del rame, metallo essenziale, importante cofattore di molte
proteine. L'incidenza è stimata essere di 1/50.000-1/80.000. Il gene della
malattia codifica per una adenosina trifosfatasi 7B (ATP7B) espressa primariamente
dal fegato, ma anche da cervello, rene e placenta. Le mutazioni causa della malattia
conducono ad un difetto della escrezione del rame nella bile (via principale di
escrezione del metallo dal nostro organismo).
![]() Omeostasi del rame: Il rame, assunto con la dieta, è assorbito dagli
enterociti prevalentemente del duodeno e del piccolo intestino prossimale ed è
trasportato nel circolo portale legato all'albumina. Il fegato lo rimuove poi
avidamente dal circolo. Il fegato utilizza una parte del rame a scopi
metabolici, sintetizza e secerne la ceruloplasmina, proteina di trasporto del
rame ed elimina il rame in eccesso con la bile. Il rame è importato all'interno
dell'epatocita, attraverso i trasportatori del rame 1 e 2 e poi consegnato al
suo sito di utilizzo attraverso un gruppo di proteine chiamate "copper
chaperones". L'ATP7B gioca due ruoli principali: uno è l'incorporazione del
rame nell'apoceruloplasmina (precursore della ceruloplasmina) convertendola a
ceruloplasmina al polo sinusale della membrana dell'epatocita; l'altro ruolo è indirizzare
il metallo al polo biliare in caso di eccesso di rame.
Omeostasi del rame: Il rame, assunto con la dieta, è assorbito dagli
enterociti prevalentemente del duodeno e del piccolo intestino prossimale ed è
trasportato nel circolo portale legato all'albumina. Il fegato lo rimuove poi
avidamente dal circolo. Il fegato utilizza una parte del rame a scopi
metabolici, sintetizza e secerne la ceruloplasmina, proteina di trasporto del
rame ed elimina il rame in eccesso con la bile. Il rame è importato all'interno
dell'epatocita, attraverso i trasportatori del rame 1 e 2 e poi consegnato al
suo sito di utilizzo attraverso un gruppo di proteine chiamate "copper
chaperones". L'ATP7B gioca due ruoli principali: uno è l'incorporazione del
rame nell'apoceruloplasmina (precursore della ceruloplasmina) convertendola a
ceruloplasmina al polo sinusale della membrana dell'epatocita; l'altro ruolo è indirizzare
il metallo al polo biliare in caso di eccesso di rame.
Sono state identificate oltre 200 mutazioni del gene ATP7B che rendono conto della variabilità dell'esordio e del quadro clinico.
Caratteristiche cliniche: La malattia di Wilson può esordire in maniera molto variabile. È ampiamente accettato che i sintomi non si sviluppino per lo meno prima dei 3 anni di età e raramente diventino evidenti prima dei 5 anni. I possibili segni e sintomi associati con WD sono menzionati di seguito.
Le principali manifestazioni cliniche riguardano, in ogni caso, il fegato ed il SNC. Sulla base dei dati della letteratura, la maggior parte dei pazienti pediatrici affetti da WD manifesta il coinvolgimento epatico, mentre sintomi neuropsichiatrici sono più comuni in pazienti più adulti.
Diagnosi: La diagnosi di malattia di Wilson è una diagnosi impegnativa, che richiede sia indagini di laboratorio, che istologiche e spesso molecolari. Nel 2003 Ferenci at al. hanno proposto un sistema diagnostico a score per la WD, che tiene in considerazione parametri clinici, laboratoristici, istologici e molecolari (ai quali viene attribuito un punteggio).
 Un'indagine di primo livello è la concentrazione
plasmatica di ceruloplasmina. La ceruloplasmina sierica è ridotta in pazienti
con WD, a causa della sua biosintesi anomala e della breve vita media delle
molecole di apoceruloplasmina libera da rame; il valore di ceruloplasmina
sierica considerato diagnostico per WD è < 20 mg/dl. Altra indagine di I
livello è la cupremia (che, però, non rientra nello score).
Un'indagine di primo livello è la concentrazione
plasmatica di ceruloplasmina. La ceruloplasmina sierica è ridotta in pazienti
con WD, a causa della sua biosintesi anomala e della breve vita media delle
molecole di apoceruloplasmina libera da rame; il valore di ceruloplasmina
sierica considerato diagnostico per WD è < 20 mg/dl. Altra indagine di I
livello è la cupremia (che, però, non rientra nello score).
Trattamento: La malattia di Wilson presenta una rapida evoluzione ed è inevitabilmente fatale se non trattata.
L'obiettivo del trattamento è prevenire l'accumulo di rame nei tessuti.
![]() L'intake di rame con la dieta deve essere limitato,
limitando i cibi ricchi in rame (come ad es. il fegato, la cioccolata, i frutti
di mare (shellfish), i funghi (mushrooms), noci).
L'intake di rame con la dieta deve essere limitato,
limitando i cibi ricchi in rame (come ad es. il fegato, la cioccolata, i frutti
di mare (shellfish), i funghi (mushrooms), noci).
È necessario un trattamento farmacologico long-life e molti farmaci sono generalmente impiegati. Il loro meccanismo d'azione è la chelazione del rame in eccesso e la riduzione del suo assorbimento. Tra i farmaci ricordiamo: la D-Penicillamina (il più importante agente chelante, gravata però da numerosi effetti collaterali) ed i sali di zinco.
Ipertransaminasemia in corso di malattia celiaca
Il coinvolgimento epatico in un paziente celiaco può presentare le caratteristiche di:
Epatite glutine-dipendente da alcuni definita "epatite celiaca"; si tratta di una epatite reattiva aspecifica, cronica, che addirittura potrebbe evolvere (come ogni epatopatia cronica) verso una insufficienza epatica e necessitare di trapianto (OLT). La dieta aglutinata ne determina la risoluzione;
Epatite autoimmune steroido-dipendente una vera e propria patologia autoimmune, che non regredisce con la dieta aglutinata, ma solo con il trattamento corticosteroideo.
Il trattamento steroideo viene iniziato nelle primi fasi con il dosaggio pieno (pari a 2 mg/kg) di prednisone e può essere associato a 1 mg/kg di azatioprina. Questo dosaggio dovrebbe spegnere la malattia e viene effettuato per due settimane. Successivamente si riducono le dosi di prednisone a 1,75-1,50 mg/kg fino ad arrivare a 5mg al dì, o a giorni alterni, in modo tale da non interferire con il metabolismo calcio-fosforo e quindi con l'altezza del paziente. Si evitano così gli effetti collaterali della terapia con corticosteroidi: cataratta, malattia di Cushing, osteoporosi, ipertensione arteriosa, ecc Se la terapia viene sospesa prima che il paziente effettui una biopsia epatica che dimostri che la malattia è spenta dal punto di vista istologico e prima che siano passati 2 anni di completa normalizzazione del bilancio epatico, la recidiva si manifesta nel 95% dei casi. Se la malattia recidiva, la terapia deve essere effettuata a dosaggi elevati. La terapia è importante in quanto circa il 60% di questi pazienti ha un quadro cirrogeno e se la diagnosi è tardiva il paziente deve effettuare un trapianto epatico.
Nei pz. celiaci, in seguito al vaccino anti-HBV, gli anticorpi anti-epatite B possono risultare scarsamente positivi perché nella malattia celiaca c'è un'interferenza con la produzione di Ab anti-HBs, quando i pazienti sono a dieta con il glutine. Il paziente in trattamento con dieta priva di glutine ha di nuovo la capacità di produrre gli Ab anti-HBs. E' buona norma fare un richiamo della dose anticorpale.
MICI e Colangite Sclerosante
Il coinvolgimento epatico in corso di MICI è quello di una colangite sclerosante primitiva. La colangite sclerosante si caratterizza per infiammazione e fibrosi concentrica (onion skin fibrosis) progressiva di segmenti delle vie biliari intra ed extraepatiche che possono:
esitare in lesioni a corona di rosario, in cui a segmenti stenosanti si alternano a segmenti risparmiati o dilatati;
oppure evolvere fino all'obliterazione completa.
Patogenesi: meccanismi immunomediati, esposizione epatica a citochine prodotte a livello dell'intestino che raggiungono il fegato attraverso il circolo portale; produzione di autoanticorpi a livello epatico e riduzione di cellule T-suppressor.
Diagnosi
esami di screening di funzionalità epatica;
positività degli ANCA;
colangio-RM (per valutare l'aspetto a corona di rosario);
la diagnosi definitiva richiede la biopsia epatica.
È stata riscontrata una nuova entità nel bambino, definita colangite autoimmune, in cui ad un quadro di colangite sclerosante si associa un quadro di epatite autoimmune (SINDROME DA OVERLAP). Non vi è associazione con MICI. I farmaci immunosoppresori sono il caposaldo della terapia.
IEF: Intolleranza Ereditaria al Fruttosio
Incidenza
Difetto enzimatico: fruttosio-1-fosfato Aldolasi B.
Difetto molecolare: ALDOB (9 q21.3-q22.2). A149P (67%), A174D (16%).
Presentazione Clinica: L'accumulo intracellulare di fruttosio-1-fosfato determina ipoglicemia, vomito ed importante coinvolgimento epatico e renale.
Esposizione Acuta: sudorazione, tremori, ipoglicemia, nausea, vomito, apatia, letargia coma, convulsioni.
Esposizione Cronica: vomito, diarrea, anoressia, ritardo di crescita, apatia, epatomegalia, ittero, coagulopatia, proteinuria.
Reperti di laboratorio: tempo di coagulazione prolungato, ipoalbuminemia, aumento dei livelli di bilirubina e transaminasi, disfunzione tubulare prossimale. I bambini affetti si proteggono mediante il rifiuto del fruttosio e possono essere asintomatici fino all'età adulta. In seguito all'ingestione di tracce di fruttosio possono presentare ipertransaminasemia e steatosi.
Terapia: dieta priva di fruttosio, saccarosio e sorbitolo, con l'integrazione di Vitamina C.
Glicogenosi

Figura : Tabella riassuntiva delle glicogenosi.
Epatopatie da Farmaci
Le epatopatie da farmaci possono essere causate da:
ANTIMICROBICI: antibatterici, anti-tubercolotici, antivirali, antifungini;
PARACETAMOLO (una somministrazione eccessiva e prolungata di paracetamolo associata ad una riduzione dell'apporto calorico o proteico può associarsi ad epatotossicità nei bambini) e FANS;
ANTI-CONVULSIVANTI;
ANTI-ACIDI (H2-antagonisti, IPP);
IMMUNOSOPPRESSORI;
CITOSTATICI.

Figura : Patter clinico-patologici e istopatologici di danno da farmaci.
La corretta anamnesi del paziente è fondamentale. Il danno epatico può essere l'unica manifestazione oppure può associarsi a manifestazioni sistemiche. Le manifestazioni cliniche possono essere lievi e aspecifiche oppure severe. Le caratteristiche di laboratorio sono estremamente variabili. Lo screening tossicologico su campioni di sangue e di urine può essere di aiuto.
Diagnosi di Esclusione: Anamnesi Accurata.
La diagnosi viene effettuate mediante uno score che prevede: la valutazione della latenza tra l'esposizione al farmaco e la reazione tossica, l'intervallo tra la sospensione/reazione tossica, intervallo tra sospensione e la normalizzazione dei parametri di laboratorio, la presenza di manifestazioni extra-epatiche con rush, febbre, artralgie, eosinofilia e citopenia, esclusione di altre cause, l'esposizione accidentale al farmaco (es. si può verificare quando il bimbo non può assumere l'augmentin - amoxicillina e acido clavulanico - e la madre quindi gli dà un "altro" antibiotico: il clavulin = amoxicillina acido clavulanico!!!).
Macro-AST
Una possibile causa di incremento isolato delle AST può essere la presenza nel siero di macro-AST: macromolecole composte da IgG legate all'AST. Questi macrocomplessi non vengono più clearati a livello renale e persistono nel siero.
Per la diagnosi: se cimentiamo in una provetta il siero del paziente con il PEG (poli-etilen-glicole), che fa precipitare le macromolecole, dopo averlo centrifugato, possiamo notare nel sottonatante i macrocomplessi, mentre nel sovranatante le transaminasi normali.
Per cui, facendo il dosaggio nel sovranatante e nel pellet (sottonatante), possiamo valutare che nel sovranatante ci sono poche transaminasi e nel sottonatante ce ne sono la maggior parte.
La diagnostica può essere affinata facendo una elettroforesi, possiamo valutare che la macro-AST (macromolecola) ha una velocità di migrazione intermedia tra l'AST citosolica e l'AST di tipo microsomiale.
Si deve sospettare la presenza di macro-AST quando abbiamo un ipertransaminasemia limitata alle AST.
Congenital Disorders of Glycosylation (CDG)
Sono un gruppo di oltre 20 patologie ereditarie che compromettono l'N-glicosilazione delle proteine. Le manifestazioni cliniche delle CDG differiscono tra loro lievemente e ciò ne rende difficile il riconoscimento.
Segni e Sintomi principali: ritardo psico-motorio, atassia, crisi epilettiche, retinopatia, steatosi/fibrosi epatica, coagulopatie, ipoglicemia, FTT, capezzolo invertito, cuscinetti di grasso sottocutaneo, strabismo. Perdita intestinale di proteine (enteropatia proteino-disperdente).
Ittero e Colestasi nel lattante
L'ittero è una condizione caratterizzata da un aumento delle concentrazioni di bilirubina nel siero che determina a livello di cute, sclere e mucose un caratteristico colorito giallastro. Tale condizione per svariati motivi è molto più frequente nel neonato rispetto al bambino più grande e ciò determina un continuo monitoraggio dei livelli di bilirubina nelle prime 48-72 ore ad intervalli di tempo di 6-12 ore o addirittura minori in caso di valori particolarmente elevati.
Questo problema neonatale necessità di un'integrazione interdisciplinare tra più clinici: ematologo e il neonatologo ad esempio se si stratta di un'iperbilirubinemia indiretta, l'epatologo viceversa se si tratta di iperbilirubinemia diretta.
Si distinguono:
Itteri a bilirubina non coniugata (indiretta);
Itteri a bilirubina coniugata (diretta).
Itteri a Bilirubina Non Coniugata
Ittero fisiologico: Condizione che coinvolge circa il 30% dei neonati caratterizzata da un'elevata produzione di bilirubina (nel neonato la velocità di produzione di bilirubina è significativamente più elevata rispetto all'adulto: 6-8 mg/24h contro 3-4 mg/24), ciò è dovuto al fatto che il neonato nasce con un numero elevato di globuli rossi (fino a 6 milioni) e per giunta questi sono più fragili, si determina quindi un'elevata emocateresi a cui consegue l'iperbilirubinemia non coniugata.
Il più delle volte (soprattutto nei neonati prematuri ma anche in quelli a termine) l'istaurarsi dell'iperbilirubinemia è favorito dall'immaturità dei processi di uptake, coniugazione ed escrezione epatica che ne rallentano lo smaltimento e da un aumento del circolo enteroepatico (la stessa molecola di bilirubina compie più volte il circolo enteroepatico prima di essere allontanata).
Affinchè l'ittero possa essere definito fisiologico devono verificarsi le seguenti condizioni:
Comparsa nel 2°-3° giorno;
Picco massimo 10-12 mg/dl;
Scomparsa entro 5°-6° giorno;
Velocità di accumulo della bilirubina <5 mg/dl/die.
In genere nei nati pretermine l'ittero fisiologico ha caratteristiche leggermente diverse in quanto compare più tardi (3°-4° giorno), raggiunge livelli leggermente maggiori (15 mg/dl) e scompare più tardi (7°-9° giorno).
Ittero patologico
Quando l'ittero non segue il decorso sopra elencato allora esso viene considerato patologico ed è necessario attivarsi sia in termini di terapia che di diagnosi dell'eziologia.
Ittero da latte materno: Il latte materno può contribuire ad accentuare l'ittero fisiologico attraverso due meccanismi ipotizzati: l'aumento del ricircolo enteroepatico e l'elevata concentrazione di acidi grassi insaturi che inibirebbero la glucuronil-trasferasi. In questo caso il comportamento dell'ittero è molto tipico, infatti la sospensione dell'allattamento consente lo sblocco dell'attività di coniugazione e la discesa dei livelli di bilirubina, i quali restano stabili anche in caso di ripresa dell'allattamento.
Disordini genetici del metabolismo della bilirubina
Sindrome di Gilbert
Condizione di ittero benigno esacerbata da stress psico-fisico dovuta ad una ridotta espressione della glucuronil-trasferasi. A livello genetico l'alterazione coinvolge il promoter del gene dell'enzima dove alcune sequenze TA (TATA box) vengono ripetute 7 volte anziché 6.
La sindrome di Gilbert, descritta per la prima volta nel 1901 dal gastroenterologo Augustin Nicolas Gilbert e colleghi, è una patologia benigna del fegato che si manifesta con iperbilirubinemia, spesso nel secondo decennio di vita. Ne è affetto circa il 7-8% della popolazione adulta ed è a carattere ereditario, trasmessa con modalità sia autosomico recessiva che dominante.
Nella sindrome di Gilbert il fegato non capta adeguatamente la bilirubina a causa di un deficit parziale di una proteina detta ligandina, che funge da proteina vettrice per l'entrata della bilirubina non coniugata all'interno nel fegato. La coniugazione della bilirubina la rende idrosolubile, dopodiché viene escreta nella bile in corrispondenza del duodeno. Nei pazienti affetti da sindrome di Gilbert la bilirubina rimane quindi parzialmente in circolo e non viene espulsa.
Inoltre, è presente un deficit parziale dell'attività dell'enzima UDP-glucoronil transferasi che coniuga una o due molecole di acido glucoronico alla molecola di bilirubina aumentandone l'idrosolubilità. L'entità di tale deficit, compreso tra il 20 e il 70% del valore normale, determina la diversa espressione clinica e gravità sintomatologica (ittero) della malattia.
Ciò comporta livelli di bilirubina generalmente di poco sopra la norma che possono aumentare momentaneamente in condizioni quali: digiuno, semidigiuno, ingestione di alcol, stress, febbre, infezioni, aumento dell'attività fisica. Se la quantità di bilirubina è elevata (maggiore di 2.5 mg/dl), si può manifestare l'ittero (colorazione gialla) della pelle e delle sclere (la parte bianca degli occhi).
Siccome la bilirubina ha una grande affinità per le strutture lipidiche della cellula, tende ad accumularsi nel tessuto nervoso, dove queste sono presenti in grandi quantità. Essendo tossica, tuttavia, a livelli elevati può determinare una parziale degenerazione delle cellule nervose con conseguente diminuzione delle prestazioni intellettuali.
Nel paziente con sindrome di Gilbert anche la produzione di insulina è leggermente superiore alla norma e non costante nell'arco della giornata. Ciò può determinare una carenza di nutrimento alle cellule nervose, contribuendo così a una depressione del tono neuro-umorale.
Manifestazioni cliniche: Un terzo dei pazienti affetti dalla sindrome sono completamente asintomatici, mentre i rimanenti due terzi accusano disturbi aspecifici quali dolori addominali, affaticabilità, cefalee e malessere.
Diagnosi: In genere si può parlare di sindrome di Gilbert quando si è in presenza di un aumento sostanziale della 'bilirubina indiretta' (valori normali inferiori a 1 mg/100 ml) e di un lieve o inesistente aumento della 'bilirubina diretta' (valori normali inferiori a 0,5 mg/100 ml). Gli esami si effettuano a digiuno. Importante è la diagnosi differenziale in caso di ittero o subittero con altre malattie epatiche che si manifestano con iperbilirubinemia. Ciò è possibile con semplici esami di laboratorio in quanto la S. di Gilbert non comporta danni funzionali al fegato, che quindi non presenterà ALT, AST, GGT alterati, la sintesi proteica epatica (valutabile mediante PT, PTT, albuminemia, protidemia) sarà nella norma, l'emocromo non presenterà eritrocitopenia (da possibile emolisi).
Un metodo economico per diagnosticare il Gilbert consiste nel test del digiuno. Se dopo una giornata di ridotto apporto calorico (una fetta di pane, una fettina di carne e un po' di frutta) la bilirubina sale, mentre tutti gli altri parametri restano nei range di riferimento è suggestivo di Gilbert. Sono comunque disponibili test genetici per fare diagnosi molecolare di Gilbert. La trasmissione è autosomica recessiva quindi parenti affetti possono orientare la diagnosi.
Terapia: Non esiste alcun tipo alcun tipo di terapia in quanto si tratta di un'alterazione benigna che solo in basse percentuali può influire sulla vita dei soggetti. Nonostante questo è da segnalare che la somministrazione di barbiturici può attenuare la sintomatologia itterica, aumentando l'attività dell'UDP glucoronil transferasi. Tuttavia, la sindrome di Gilbert riduce la capacità del fegato di detossificare determinati farmaci. Per esempio, la sindrome di Gilbert è associata a forte diarrea e neutropenia in pazienti che assumono irinotecano, che viene metabolizzato da UGT1A1 (glicuroniltransferasi).
Sindome di Crigler-Najjar tipo I-II
In questo caso la mutazione interessa la regione codificante dell'enzima con conseguente produzione di una glucuronil-trasferasi poco attiva.
La forma di tipo I è molto più grave e determina accumulo di bilirubina a livello dei gangli della base, vanno subito trattati con fototerapia ed exsanguino trasfusione e successivamente richiedono il trapianto di fegato (la percentuale di successo del trapianto di fegato è ormai molto alta).
La forma di tipo II è più lieve, determina un ittero moderato compatibile con la vita.
L'analisi della bile tramite HPNC è un ottimo strumento diagnostico e si basa sulla valutazione dei livelli di bilirubina monoconiugata e diconiugata:
Tipo I: bilirubina del tutto non coniugata;
Tipo II: tracce di bilirubina diconiugata e monoconiugata.
La somministrazione di Fenobarbital, che induce iperplasia del reticolo endoplasmatico e del Golgi degli epatociti con aumento dell'attività enzimatica, è un altro valido strumento per fare diagnosi differenziale tra le due forme: nel tipo II il trattamento permette il miglioramento clinico ed un controllo eccellente dei livelli di bilirubina, nel tipo I la terapia è inefficace a causa della completa assenza dell'enzima e per la produzione di un enzima inefficace.
La fototerapia non può essere perseguibile con l'aumentare dell'età a causa dell'ispessimento della cute che ostacola il passaggio dei raggi.
I test molecolari che possono farci risalire al tipo di mutazione a monte della patologia non sono tuttavia utili dal punto di vista clinico nella differenziazione tra le due forme.
Altre cause di ittero a bilirubina non coniugata neonatale
Emolisi: valutazione livelli di emoglobina, morfologia globuli rossi e reticolociti, non aptoglobina. Nel momento in cui si è certi che si tratti di un'anemia emolitica congenita si procede verso la caratterizzazione, ad esempio si può valutare l'attività della G6PD, della PK (anemia emolitica da deficit enzimatico) o la forma degli stessi globuli rossi (sferocitosi, ellissocitosi);
Sepsi
Ematomi interni o esterni (in corso di riassorbimento dell'ematoma);
Ipoalbuminemia (la bilirubina viene veicolata nel sangue dall'albumina);
Farmaci;
Ipertransaminasemia;
Ipotiroidismo;
Ostruzioni intestinali alte.
Complicanza dell'ittero patologico non trattato: il Kernittero
Il Kernittero, definito anche encefalopatia da bilirubina è una sindrome neurologica secondaria al deposito di bilirubina indiretta nei gangli della base e nei nuclei del tronco encefalico. In genere livelli di bilirubina indiretta superiori a 30 mg/dl pongono il neonato ad alto rischio di sviluppare questa terribile complicanza. La tossicità della bilirubina sembra correlata alla sua capacità di danneggiare la membrana cellulare e interferire con l'utilizzo dell' ossigeno a livello cerebrale.
I Sintomi iniziali comprendono inappetenza, perdita di riflesso del Moro e letargia, in seguito possono comparire ipertonia dei muscoli estensori, opistono e convulsioni; la maggior parte di questi neonati muoiono, quelli che sopravvivono sono seriamente compromessi dal punto di vista neurologico e presentano disturbi del movimento, sguardo verso l'alto e sordità neurosensoriale.
Terapia
Fototerapia: è indicata per livelli di bilirubina tra i 15 e i 20 mg/dl dal terzo giorno di vita in poi o anche minori in caso di comparsa precoce dell'ittero (entro le prime 24 ore). Ha l'obbiettivo di rendere la molecola più polare tramite la una fotoisomerizzazione e di facilitarne così l'allontanamento dall'organismo. Per ottenere questo si utilizza la luce dello spettro del blu (lunghezza d'onda 420 -470 nm) che viene assorbita al massimo dalla bilirubina. Tale reazione permette la conversione dell'isomero 4z-15z in altre 2 molecole: l'isomero 4z-15E (escreto con la bile) e la lumirubina che viene escreta attraverso il rene. Durante il trattamento il bambino va girato spesso per permettere alla luce di agire sull'intera superficie del corpo. In corso di fototerapia è possibile osservare alcune complicanze: sindrome del neonato di bronzo, rash maculo papuloso, porfirinemia transitoria, feci poco formate, surriscaldamento e disidratazione;
Exsanguino trasfusione: in quelle condizioni in cui l'ittero è molto elevato e può esitare nell'accumulo di bilirubina a livello dei gangli della base (Kernittero);
Fenobarbital (trattamento farmacologico): da somministrare solo in caso siano presenti deficit enzimatici (vedi sindrome di Crigler-Najjar);
Trapianto epatico: scelta terapeutica salvavita in caso di Crigler-Najjar tipo I.
Se l'ittero è secondario ad atre cause la risoluzione dell'iperbilirubinemia prevede il trattamento delle cause sottostanti (antibioticoterapia per la sepsi, drenaggio dell'ematoma, ecc.).
Ittero a bilirubina coniugata e colestasi
Un aumento della bilirubina coniugata nel sangue che si protrae per più di 14 giorni dalla nascita coincide con una condizione definita colestasi. Se l'aumento della bilirubina non coniugata nel sangue può essere fisiologico, l'aumento della bilirubina coniugata è sempre patologico.
Per colestasi si intende la riduzione del flusso biliare canalicolare; può essere dovuta a cause infettive, genetiche, metaboliche o indeterminate che danno luogo ad un'ostruzione meccanica al flusso biliare o ad un'alterazione funzionale della secrezione biliare o della funzione escretrice epatica. Tale condizione si manifesta in 1 ogni 2500 nati.
I segni clinici che orientano verso un ittero da colestasi sono l'osservazione di feci chiare o ipocoliche e urine scure (in genere le urine del neonato sono incolori) insieme ad un certo grado di epatomegalia.
Il ridotto flusso biliare determina a monte (sangue e tessuti) l'accumulo di tutte le sostanze escrete, normalmente, con la bile:
Sali biliari che più tardivamente possono dar luogo a prurito;
Il colesterolo che può accumularsi a livello cutaneo formando xantomi soprattutto a livello delle pieghe;
Bilirubina coniugata che determina ittero.
La stasi di queste sostanze a livello duttale ed epatocitario a lungo andare esita in un quadro di atresia biliare, fibrosi e degenerazione nodulare che determina l'insorgenza di ipertensione portale con tutte le complicanze ad essa associata.
A valle la riduzione dell'escrezione di bile nell'intestino oltre ad influenzare il colore delle feci può portare ad un malassorbimento di grassi e vitamine liposolubili.
Le colestasi molto schematicamente possono essere classificate in:
colestasi extraepatiche;
colestasi intra-extraepatiche;
colestasi intraepatiche.

Figura : Cause principali di colestasi.
Atresia delle vie biliari
L'atresia delle vie biliari (AVB) è un processo necroinfiammatorio progressivo, che coinvolge parzialmente o totalmente l'albero biliare extraepatico. La AVB è la causa più comune di ittero colestatico neonatale con una prevalenza di 1 ogni 10.000-15.000 nati vivi, cioè circa 700 neonati affetti ogni anno in Europa. La malattia esordisce nei primi giorni di vita e in genere evolve verso l'obliterazione dei dotti biliari extraepatici, l'interruzione del flusso biliare, la colestasi e il danno epatico cronico. Nel 20% dei casi l'atresia si associa ad un'altra malformazione, la polisplenia.
Ne esistono fondamentalmente due varianti:
In un primo caso è possibile un coinvolgimento dei soli dotti segmentali distali mentre le vie biliari extraepatiche sono pervie fino all'ilo. Questo tipo è più facilmente correggibile ma rappresenta una minoranza dei casi;
In un secondo caso (95% circa) il coinvolgimento riguarda le vie biliari extraepatiche, in base alle vie biliari coinvolte si distinguono tre tipi di atresia:
tipo I (coledoco);
tipo II (coledoco, cistico ed epatico comune);
tipo III (dotti epatici destro e sinistro), che rappresenta la forma più grave e più frequente.
Eziologia: L'origine e la causa della malattia sono ancora discusse: l'agente eziologico sembra essere rappresentato da un fattore lesivo variabile e spesso non precisabile che, attraverso un meccanismo infiammatorio immunomediato, agirebbe su vie biliari in precoce formazione, determinandone l'atresia.
Diagnosi
Anamnesi ed esame obiettivo: I neonati con AVB presentano età gestazionale e peso alla nascita normali con accrescimento regolare nelle prime settimane di vita, malgrado l'ittero persistente ed ingravescente, accompagnato da acolia fecale costante per oltre 8 gg consecutivi, epatomegalia ed aumento di consistenza del fegato progressivi;
Esami di laboratorio: Evidenziano un quadro di colestasi (bilirubina diretta > 50% della totale, SGOT, SGPT e GT 2-3 volte la norma);
Esami strumentali: L'ecografia epatobiliare è la metodica strumentale non invasiva di scelta, poiché può dimostrare il caratteristico segno del triangular cord (massa fibrosa a forma triangolare situata cranialmente alla biforcazione della vena porta). Di recente è stato proposto un altro segno (gallbladder ghost triad) rappresentato dal riscontro di colecisti atresica di lunghezza inferiore a 1,9 cm, dismorfica, a contorno irregolare, con perdita dell'ecogenicità della mucosa e spessore indistinto della parete. L'aumento di volume del fegato, l'ecostruttura diffusamente disomogenea ed iperecogena, la splenomegalia e l'eventuale riscontro di polisplenia sono segni indicativi complementari. La scintigrafia epatobiliare con derivati dell'acido iminoacetico marcati con tecnezio 99 è utile per distinguere l'atresia da una colestasi non ostruttiva, infatti nell'atresia a causa dell'obliterazione dei dotti la captazione epatica è normale mentre quella intestinale è assente.
In caso di sospetto clinico ed ecografico l'iter successivo comprende la laparotomia esplorativa e la biopsia epatica che permette l'esame istologico il quale può mostrare il reperto caratteristico di:
Proliferazione dei duttili;
Presenza di tappi ed edema portali e periportali con associata fibrosi portare e periportale.
A differenza dell'epatite neonatale l'architettura epatica è conservata.
Terapia: Consiste in una derivazione biliodigestiva, rappresentata dall'epatico-digiuno anastomosi, nel caso di atresia di tipo I e II, oppure dalla epatoportoenterostomia secondo Kasai nel caso di atresia di tipo III. L'intervento prevede l'asportazione completa delle vie biliari extraepatiche atresiche con anastomosi all'ilo biliare epatico di un'ansa intestinale montata a Y, allo scopo di consentire il drenaggio dai canalicoli biliari intraepatici neoformati a livello dell'ilo epatico (porta Hepathis).
Il razionale di questo intervento è che possano essere presenti nella compagine del tessuto fibroso dell'ilo epatico dei dotti biliari residui che sono posti a drenare nell'intestino la bile presente nell'intero sistema duttale intraepatico.
Il successo dell'intervento dipende da alcuni paramentri:
L'esperienza dell'ospedale dove viene effettuato l'intervento;
La tempistica dell'intervento: infatti il flusso si ristabilisce in un numero maggiore di casi quando l'intervento è effettuato precocemente ed anche i risultati a lungo termine sembrano essere migliori;
La presenza di duttili di diametro maggiore di 150 micrometri è associata ad una maggiore percentuale di successo.
L'intervento di Kasai spesso non è risolutivo ma permettere di posticipare la cirrosi e di permettere quindi una crescita normale al neonato prima che vi sia la possibilità di un trapianto epatico.
La ricomparsa dell'ittero dopo intervento di Kasai è frequente (40-60% nelle varie casistiche) e la percentuale di pazienti anitterici guariti a distanza di oltre 10 anni, risulta essere attorno al 20%. La causa più probabile è rappresentata dalla persistenza di un processo infiammatorio, sostenuto dall'incremento di molecole chemotattiche o di adesione cellulare e dall'attivazione di cellule infiammatorie perisinusoidali in fibroblasti, con conseguente aumento di sintesi e deposito di collagene, che provocherebbe la progressiva ed irreversibile trasformazione cirrotica del fegato, nonostante la disostruzione chirurgica.
Colestasi intraepatica
Fino ad una quarantina di anni fa tra le cause di colestasi neonatale venivano annoverate forme virali, l'atresia delle vie biliari e poi esisteva una forma denominata "epatite neonatale" che rappresentava un grande calderone in cui venivano riunite una serie di epatopatie ad impronta colestasica ad incerta eziologia.
Oggi grazie allo sviluppo della biologia molecolare e a tecniche diagnostiche d'avanguardia è stato possibile una caratterizzazione più accurata, la tabella seguente illustra schematicamente ed in maniera ordinata queste diverse forme.
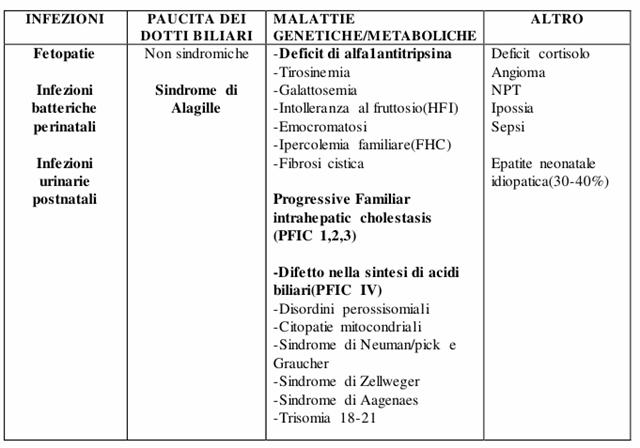
Figura : Cause di colestasi intra-epatiche.
I fattori che devono far pensare ad una colestasi intraepatica sono:
La familiarità per forme ad eziologia genetica;
Peso basso alla nascita;
L'ipossia.
Fetopatie: Se la mamma durante la gravidanza contrae un infezione da parte di microrganismi del complesso TORCHESL (toxoplasma, rosolia, citomegalovirus, herpes simplex, sifilide, listeria) tra i tanti disturbi vi può essere un coinvolgimento epatico con epatite ad impronta colestasica.
Infezioni batteriche perinatali e infezioni urinarie: Alcune infezioni sistemiche ed in particolare quelle delle vie urinarie (Escherichia coli) si associano molto spesso all'insorgenza di colestasi intorno al 15 giorno di vita, il meccanismo patogenetico non è del tutto chiarito ma si pensa che alcuni batteri siano in grado di produrre tossine ad azione colestatogena.
Disturbi del trasporto, della secrezione, della coniugazione e della biosintesi degli acidi biliari: Le PFIC (Progressive Familiar intrahepatic cholestasis) sono delle patologie in cui il difetto di proteine, implicate nel trasporto canalicolare degli acidi biliari o nella loro sintesi, determinano l'insorgenza di colestasi ad andamento progressivo, se ne conoscono 4 forme:
la PFIC tipo I o morbo di Byler;
La PFIC tipo II o sindrome di Byler;
La PFIC tipo III;
La PFIC tipo IV in cui rientrano difetti di sintesi.
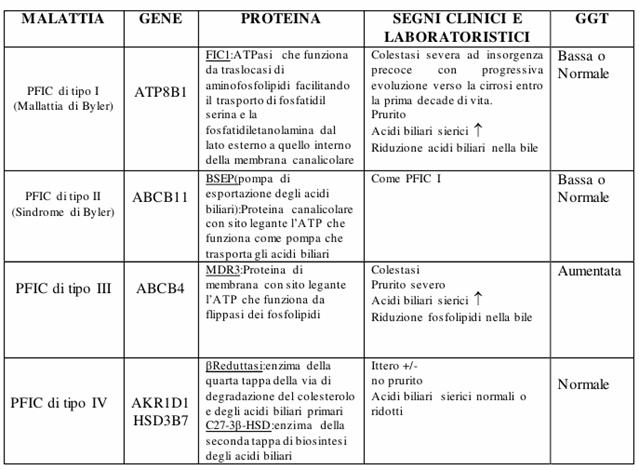
Figura : Tabella con le principali PFIC.
Accanto a queste forme più severe elencate in tabella esistono le BRIC (Colestasi Intraepatica Benigna Ricorrente) caratterizzate da picchi ricorrenti di colestasi, ittero e prurito intenso che durano da 2 settimane a 6 mesi; in queste forme la progressione verso la cirrosi è rara, probabilmente il deficit meno severo del trasportatore è responsabile della sintomatologia clinica più lieve di queste malattie.
L'alterato trasporto di acidi biliari o fosfolipidi determina colestasi in quanto per una densità ottimale della bile è necessario un perfetto equilibrio tra fosfolipidi ed acidi biliari. Dal punto di vista clinico le PFIC di tipo I e II sono molto simili e si differenziano oltre che per il tipo di gene coinvolto anche perché dal punto di vista storico le prime sono state descritte per la prima volta nelle famiglie Amish mentre le seconde in famiglie non Amish (Soprattutto europee).
Alla microscopia elettronica la bile nella PFIC I appare granulare a differenza della bile nella PFIC tipo II che risulta essere amorfa. Le PFIC tipo I e II presentano alti livelli sierici di acidi biliari ed una GGT normale o ridotta, ciò differenzia queste due forme dalla PFIC di tipo III dove la GGT è aumentata.
Il contenuto normale di acidi biliari nel sangue ed una GGT normale depone invece per un difetto di sintesi degli acidi biliari.
Sindrome di Alagille o displasia arteriepatica
La sindrome di Alagille è la più frequente sindrome con paucità di dotti biliari intraepatici (1:100.000 nati), dal punto di vista istologico si osserva una netta riduzione o la scomparsa dei dotti biliari intraepatici della triade portale, con rami portali e arteriola epatica normale. Spesso la biopsia mette in evidenza un processo infiammatorio che suggerisce una distruzione immunitaria dei dotti, ma l'ipotesi più accreditata è quella di un difetto dell'embriogenesi.
Dal punto di vista genetico la sindrome sembra legata alla mutazione del gene jagged1 (cromosoma 20) codificante per il ligando di notch 1.
La paucità dei dotti biliari intraepatici è associata ad altre anomalie:
Facies caratteristica: fronte ampia, occhi infossati e distanziati, naso lungo e dritto, ipoplasia mandibolare;
Anomalie cardiache: stenosi periferica dell'arteria polmonare, tetralogia di Fallot;
Anomalie oculari: embriotoxon posteriore (anomalia congenita della cornea che presenta una stria biancastra localizzata nella membrana di Descemet e sporgente nella camera anteriore) e pigmentazione retiniche;
Anomalie scheletriche: vertebre a farfalla (mancata chiusura dell'arco anteriore delle vertebre);
Nefropatia tubulointerstiziale.
In aggiunta può essere presente un ritardo di crescita e deficit della spermatogenesi: il decorso è variabile da paucisintomatico senza itero e prurito fino a severa colestasi con ipercolesterolemia e xantomi, l'insufficienza epatica è rara (cirrosi 46%).
Deficit di α1antitripsina
L'α1antitripsina è il più potente inibitore sierico di proteasi, sono presenti più di venti alleli codominanti del suo gene, l'allele più comune è l'allele M, mentre l'allele Z predispone verso il deficit della proteina, tenuto conto di tutto questo i soggetti con genotipo ZZ presentano bassissimi livelli dell'enzima (<2mg/dl) e sono predisposti a sviluppare patologie epatiche e polmonare.
Mentre nell'adulto tale deficit è associato ad un quadro polmonare di enfisema (mancata inibizione delle elastasi neutrofile), nel bambino il deficit si associa a epatopatia colestasica neonatale e cirrosi infantile. Il fenotipo è molto variabile.
Il modello proposto vede la patogenesi associata ad un difettoso ripiegamento dell'allele z che favorisce la formazione di polimeri che sono trattenuti nel reticolo endoplasmatico (ciò è confermato dalla presenza di accumuli pas positivi all'interno delle cellule di Kuppfer, negli epatociti periportali e nei biliociti) determinando a lungo andare un danno cellulare.
La diagnosi differenziale viene effettuata attraverso la valutazione del quadro elettroforetico che mostra la riduzione della banda α-1.
Tirosinemia, intolleranza al fruttosio, emocromatosi e galattosemia
Sono malattie ad eziologia diversa accomunate però da alcune caratteristiche cliniche:
Tutte e quattro le forme esordiscono con un quadro di colestasi severa e se non diagnosticate e trattate evolvono rapidamente verso l'insufficienza epatica;
Sono associate ad una discoagulopatia con tempo di quick molto allungato.
La galattosemia è una malattia metabolica a carattere autosomico recessivo caratterizzata da un deficit nella metabolizzazione del galattosio: esistono tre forme di deficit di cui una riguarda il galattosio-1-fosfato uridiltransferasi, un'altra la galattocchinasi e l'ultima l' uridina difosfato galattosio 4-epimerasi. Ciò determina accumulo di galattosio e galattitolo a livello ematico e oculare (cataratta).
Dal punto di vista clinico abbiamo:
Grave danno epatico con colestasi;
Coinvolgimento renale;
Frequenti infezioni da escherichia coli;
Crescita stentata, diarrea, vomito;
Cataratta;
Coagulopatia severa.
Per la diagnosi è utilizzato il Clinitest, un test urinario che valuta la presenza di sostanze riducenti all'interno delle urine (potrebbe essere uno zucchero qualsiasi, galattosio, fruttosio, lo stesso glucosio).
Per una migliore caratterizzazione del tipo di sostanza si utilizza la cromatografia degli zuccheri su strato sottile: cromatografia su strato sottile o TLC, acronimo dell'inglese Thin Layer Chromatography. Come tutte le cromatografie, si basa sulla diversa ripartizione di sostanze differenti tra una fase stazionaria ed una fase mobile, in funzione dell'affinità di ogni sostanza con esse. La fase stazionaria è generalmente uno strato dallo spessore uniforme di circa 1 mm di materiale adsorbente, depositato su una lastra di vetro. Il materiale adsorbente può essere gel di silice, allumina, cellulosa in polvere o polvere di diatomee (kieselguhr), a seconda dell'applicazione richiesta. La fase mobile è un solvente opportunamente scelto (o una miscela di solventi), capace di separare i componenti della miscela da analizzare e poco affine per polarità alla fase stazionaria scelta. Uno strato di fase mobile alto circa 1 cm viene posto sul fondo di un contenitore, nel quale si immerge l'estremità inferiore della lastra di vetro preparata col materiale adsorbente, sulla quale è stata previamente posta una goccia del campione da separare (o di una sua soluzione). Il contenitore viene poi chiuso in modo da mantenere l'ambiente saturo di vapori di solvente. Per effetto di capillarità il solvente sale lungo la lastrina, trascinando con sé in maniera differente i componenti della miscela e separandoli. A questo punto avviene la colorazione che permette di mettere in evidenza le diverse bande di accumulo. Il test più sensibile resta comunque il dosaggio enzimatico dell'enzima che si sospetta sia carente.
Queste forme metaboliche rare come già detto hanno una progressione cirrogena rapida e possono esitare addirittura il noduli displastici e veri e propri epatocarcinomi, quando la cirrosi è tale da determinare insufficienza epatica si rende necessario il trapianto epatico.
Altre cause di epatopatia con colestasi
Altre cause di epatopatia con colestasi sono le malattie da accumulo (Neuman-pick, Gaucher), in queste malattie oltre alla epatomegalia è presente una splenomegalia particolarmente significativa non giustificato dal quadro epatico, lo striscio periferico di sangue ci permette di fare diagnosi attraverso l'evidenza di cellule da accumulo (cellule schiumose).
I bambini affetti da malattie mitocondriali oltre alla colestasi presentano un ipotonia marcata ed acidosi sistemica.
La nutrizione parenterale è una nota noxa colestatogena, il digiuno prolungato altera l'integrità della mucosa enterale e facilità la risalita di batteri attraverso le vie biliari inoltre la riduzione del rilascio di ormoni come CCK riduce il flusso biliare.
Riduzione del flusso biliare (colestasi) ed infezioni ricorrenti sono i principali fattori che determinano l'insorgenza del danno epatico.
|
| Appunti su: Disordini congeniti della N-glicosilazione, forsa cupremi, cos27C3A8 la colestasi intraepatica si vede con la colangiografia rm, bambini con ipotono da celiachia, hpnc ittero, |
|
| Appunti Bambini |  |
| Tesine Bellezza |  |
| Lezioni Nutrizione |  |