|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 3313 | Gradito: |
Leggi anche appunti:MastocitosiMastocitosi Sono un gruppo eterogeneo di patologie accomunate da un netto Sindrome di downSINDROME DI DOWN Disordine cromosomico che di solito comporta ritardo mentale, Patologia generalePATOLOGIA GENERALE Breve riassunto del'Infiammazione |
 |
 |
LO SVILUPPO SESSUALE E PATOLOGIE CORRELATE
La differenziazione sessuale
La determinazione genetica del sesso è correlata alla presenza o assenza del gene SRY sul cromosoma Y: in assenza del gene, infatti, lo sviluppo procede verso la differenziazione in senso femminile.
Il processo viene modificato da un fattore di determinazione del sesso codificato dal gene SRY, il quale durante la vita embrionale, intorno alla 4-5 settimana, induce la differenziazione delle cellule del Sertoli. Le cellule del prosertoli si sviluppano nei cordoni sessuali presenti nella cresta genitale. Inoltre il fattore del SRY induce la formazione delle cellule del Leydig nella midollare (ossia nel mesenchima) della cresta genitale.
Il corretto sviluppo del testicolo fa sì che esso divenga in grado di sintetizzare:
AMH o MIS, Mullerian inhibiting substance, secreta dalle Sertoli che provoca l'involuzione dei dotti di Muller;
androgeni, secreti dalle Leydig, che orientano lo sviluppo dei genitali esterni e la migrazione dei testicoli.
![]()
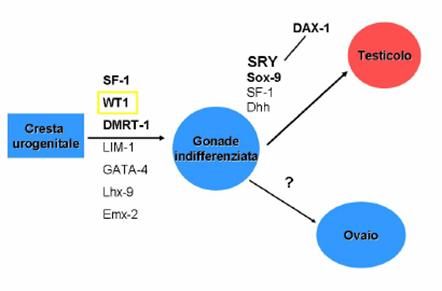 Il gene SRY si trova sul cromosoma Y, in
prossimità della regione PAR1 (pseudoautosomal region) che ha una elevata
omologia (100%) con la regione PAR1 presente sul cromosoma X; durante la
meiosi, X e Y si appaiano su tale regione e affinché si abbia una corretta
segregazione dei cromosomi X e Y, su PAR1 deve avvenire almeno un evento di
crossing over.
Il gene SRY si trova sul cromosoma Y, in
prossimità della regione PAR1 (pseudoautosomal region) che ha una elevata
omologia (100%) con la regione PAR1 presente sul cromosoma X; durante la
meiosi, X e Y si appaiano su tale regione e affinché si abbia una corretta
segregazione dei cromosomi X e Y, su PAR1 deve avvenire almeno un evento di
crossing over.
Se il crossing over avviene in modo corretto non si hanno problemi quindi un individuo XY sarà maschio e un individuo XX sarà femmina. Se il crossing over avviene in modo ineguale è possibile che si abbia la traslocazione di SRY su cromosoma X. Da tale evento si ottengono individui XX maschi e individui XY femmine. Il fenomeno prende il nome di sex reversal.
Pubertà
La pubertà è caratterizzata dalla comparsa di sviluppo dei caratteri sessuali secondari, si completa nell'arco di tre-sei anni ed è suddivisa in tappe maturative secondo vari modelli il cui più conosciuto è quello di Turner, con 5 stadi maturativi.
Si valutano: sviluppo mammario, sviluppo pilifero dei genitali e sviluppo dei genitali maschili (scroto e pene).
Lo sviluppo mammario prende l'acronimo di B o M (breast o mammella); quello pilifero dei genitali prende l'acronimo di P o PH. Quello dei genitali maschili prende l'acronimo di G.
Qual è il primo segno di pubertà? Nel maschio c'è il Gonadarca ovvero l'aumento di dimensione e di pigmentazione dello scroto. Nella donna c'è il Telarca cioè la comparsa del bottone mammario.
L'età di inizio della pubertà va dagli 8 ai 13 anni per le femmine e 9-14 per i maschi. Questo dipende anche dal quadro familiare. La pubertà viene considerata precoce prima degli 8 anni nella bambina e 9 nel maschio. È ritardata quando compare dopo i 13 anni nella femmina e dopo i 14 anni nel maschio.
La pubertà è il fenomeno evolutivo che completa l'infanzia introducendo l'individuo nell'adolescenza. È caratterizzata dalla maturazione dei caratteri primari e secondari e dal "growth spurt", ovvero l'accelerazione della crescita con cui si raggiunge la statura definitiva.
 Nelle femmine il primo segno di pubertà visibile
è la comparsa del bottone mammario tra gli 8 e i 12 anni. La mestruazione
inizia dopo 2-2,5 anni (range normale:9-16 anni). Nei maschi il primo segno
visibile di pubertà è l'ingrossamento dei testicoli a partire dai 9,5 anni; ad
esso segue la crescita del pene.
Nelle femmine il primo segno di pubertà visibile
è la comparsa del bottone mammario tra gli 8 e i 12 anni. La mestruazione
inizia dopo 2-2,5 anni (range normale:9-16 anni). Nei maschi il primo segno
visibile di pubertà è l'ingrossamento dei testicoli a partire dai 9,5 anni; ad
esso segue la crescita del pene.
![]() Durante l'adolescenza nella femmina gli scatti
della crescita hanno un picco a 11 anni e mezzo mentre nel maschio il picco si ha
più tardi, a 13 anni e mezzo. La stimolazione androgenica delle ghiandole
sebacee e apocrine provoca odori corporei e acne.
Durante l'adolescenza nella femmina gli scatti
della crescita hanno un picco a 11 anni e mezzo mentre nel maschio il picco si ha
più tardi, a 13 anni e mezzo. La stimolazione androgenica delle ghiandole
sebacee e apocrine provoca odori corporei e acne.
Variazioni Del Gonadostato: La pubertà inizia tra gli 8 e i 13 anni nelle femmine e i 9 e i 14 anni nei maschi. L'ipotalamo inizia a produrre una maggiore quantità di ormoni che agiscono sull'ipofisi e sulle gonadi e sono regolati mediante un meccanismo a feed-back.
I disordini puberali si suddividono in:
Telarca precoce;
adrenarca precoce;
pubertà precoce vera;
pseudopubertà precoce;
pubertà ritardata.
Telarca Precoce Isolato
Il telarca precoce isolato è caratterizzato da un aumento di volume della ghiandola mammaria unilaterale o bilaterale in una bambina di età <3 anni . Negli ultimi anni sono stati chiamati in causa anche gli alimenti ricchi di estrogeni(esempio carne di pollo, uova). L'FSH presenta valori aumentati in circolo. Assenza di altri segni di maturazione puberale: il menarca compare all'età giusta e anche la riproduzione è normale.
L'età ossea di questi pazienti corrisponde a quella cronologica con altezza finale leggermente ridotta. Se l'esposizione è continua può ritardare, in alcuni casi, l'altezza finale mentre se è parziale non lascia segno di sé. Regredisce spontaneamente.
L'adrenarca precoce
L'adrenarca precoce è caratterizzato dalla presenza di peli pubici e ascellari nei bambini di età<5-8 anni. Non ci sono altri segni di maturazione sessuale. Sono aumentati gli androgeni, il DHEA e il DHEA-S. L'età ossea è avanzata rispetto all'età cronologica. E' necessario effettuare un follow-up in quanto se il fenomeno non si autolimita può causare un ritardo di crescita. L'adrenarca è una condizione a progressione lenta e non richiede terapia.
Pubertà Precoce Vera
La pubertà precoce vera consiste nella maturazione sessuale con la comparsa di tutti i caratteri sessuali secondari nei maschi di età inferiore ai 9 anni e nelle femmine di età inferiore agli 8 anni. E' presente una prematura attivazione dell'asse ipotalamo-ipofisi-gonadi. Si manifesta con maggiore frequenza nelle femmine. Si verifica una precoce chiusura epifisaria e la statura finale è inferiore a quella che si sarebbe raggiunta altrimenti.
Cause
Tumori dell'ipotalamo;
Tumori dell'epifisi;
Patologie del SNC: traumi cranici, encefaliti, meningiti, interventi chirurgici, malformazioni, altri tumori cerebrali, radiazione, malformazioni;
Ipotiroidismo primario.
Diagnosi: aumento del 17-β estradiolo nella femmina e del testosterone nel maschio. Le concentrazioni sieriche di estradiolo nelle bambine sono basse o indosabili nella fase iniziale della precocità sessuale come nella pubertà normale.
I livelli di FSH ed LH in circolo risultano aumentati. Il test con le gondadotropine determina un aumento dell'LH. Nella pubertà precoce vera il test con il GnRH determina un aumento del FSH e l'LH appare molto aumentato. Se il bambino presenta il telarca è l'FSH che è molto aumentato.

Figura : Possibili risposte al test da carico con il GnRH (LHRH).
Esami strumentali
Rx per valutare l'età ossea;
Eco pelvica per valutare follicoli ovarici. E' utile anche per valutare il rapporto tra fondo dell'utero e corpo dell'utero e per valutare la presenza di eventuali masse pelviche;
RM e TC per escludere tumori o altre lesioni del SNC.
Terapia medica: Nella Pubertà precoce idiopatica: Somministrazione continua di analoghi (agonisti) del GnRH che desensibilizzano i siti recettoriali delle cellule gonadotrope. Quando il paziente arriva al momento fisiologico della pubertà si sospende la terapia medica (effetto soppressivo reversibile alla sospensione della terapia). Cardine della terapia è l'analogo long-acting dello LHRH ad azione agonista. In Italia vi è attualmente l'autorizzazione per la Triptorelina (poamoato e acetato) (Decapeptyl-Gonapeptyl) e la Leuprolina (Enantone). La Pubertà precoce secondaria, invece, richiede il trattamento della causa sottostante (es.tumori del SNC).
Pseudo-Pubertà Precoce
La pseudo-pubertà precoce è indipendente dagli ormoni, infatti l'LH e l'FSH sono diminuiti. Non è presente una iperattivazione dell'asse ipotalamo-ipofisi-gonade. Si manifesta prima dell'età del nono anno nei maschi e dell'ottavo anno nelle femmine. E' dovuta ad una produzione autonoma degli ormoni sessuali. Le cause di pseudo-pubertà precoce sono:
Iperplasia surrenalica congenita (s.adreno-genitale);
Tumori che secernono gonadotropine (epatomi,epatoblastomi, germinomi del SNC, corioepiteliomi);
Tumori surrenalici;
Iperplasia testicolare familiare;
Tumori dell'ovaio;
Sindrome di McCune-Albright;
Sindrome di Peutz-Jeghers.
La sindrome di Mc Cune Albright è caratterizzata da iperpigmentazioni presenti in età precoce, anomalie vascolari che determinano la pubertà precoce (il ciclo mestruale può iniziare a anche a 4-6 mesi), gigantismo, macchie caffè-latte, deformità ossee. La malattia è caratterizzata da una iperfunzione autonoma di molte ghiandole.
La sindrome di Peuthz Jeghers è caratterizzata da poliposi intestinale associata ad ipermelanosi delle labbra. La sindrome di Peutz-Jeghers è una malattia nella quale la persona sviluppa polipi intestinali ed ha un rischio significativamente più elevato di sviluppare alcuni tumori. Il National Institutes of Health stima che colpisce circa un neonato ogni 300.000. Ci sono due tipi di sindrome di Peutz-Jeghers:
Sindrome di Peutz-Jeghers ereditaria, dovuta ad una mutazione in un gene chiamato STK11. Il difetto genetico si tramanda ereditariamente con modalità autosomica dominante;
Sindrome di Peutz-Jeghers sporadica che non si trasmette ereditariamente e sembra slegata dalla mutazione del gene STK11.
Cause più frequenti di pseudopubertà precoce Isosessuale: Nei maschi:
deficit di 21-idrossilasi;
deficit di 11-idrossilasi;
residui surrenalica nel testicolo;
carcinoma surrenalico;
tumore a cellule interstiziali del testicolo;
maturazione precoce delle cellule di Leydig e germinali (testotossicosi);
ipotiroidismo;
iatrogena.
Nelle femmine:
cisti follicolari;
tumori a cellule della granulosa della teca;
residui surrenalici nell'ovaio (rari);
iatrogena;
ipotiroidismo;
snd. di McCune-Albright.
Cause più frequenti di pseudopubertà precoce Eterosessuale: Nei maschi:
carcinoma surrenale femminilizzante;
iatrogena.
Nelle Femmine:
deficit di 21-idrossilasi;
iatrogena.
Disordini dello sviluppo sessuale (in passato detti stati intersessuali)
Condizioni nelle quali l'aspetto dei genitali esterni è ambiguo o in disaccordo con il sesso cromosomico o gonadico della persona.
Sindrome di Morris o sindrome da resistenza agli androgeni: Soggetti 46, XY i quali, a causa di una mutazione sul cromosoma X a trasmissione recessiva (1/3 dei casi de novo), hanno una disfunzione del recettore degli androgeni consistente in un deficit di azione degli androgeni a livello dei tessuti bersaglio.
1:20.000 soggetti 46, XY. Poiché il gene SRY è presente e normalmente funzionante, i testicoli sono normalmente differenziati e sviluppati, mancano i genitali interni femminili (a causa dell'azione di AMH) ma a causa della mancata azione androginica i testicolo vengono ritenuti in addome e il fenotipo è femminile o solo parzialmente maschile. Si distinguono due forme cliniche:
l'insensibilità completa agli androgeni (CAI) è caratterizzata da un fenotipo femminile senza ambiguità genitale esterna;
le insensibilità parziali agli androgeni (PAI) sono molto eterogenee e si associano ad un ampio spettro clinico, dal morfotipo quasi femminile fino al maschio sterile, senza ambiguità dei genitali.
Nei casi fenotipicamente ben determinati in genere l'alterazione non viene rilevata fino alla mancata comparsa del menarca nei soggetti con fenotipo femminile, o fino alla ricerca delle cause di sterilità per i soggetti con fenotipo maschile.
La presenza di tessuto gonadico alterato e/o di testicoli ritenuti in addome aumenta di molto il rischio di cancro, pertanto è importante riconoscere e trattare chirurgicamente questi soggetti il prima possibile.
Sindrome di Swyer o disgenesia gonadica pura: Condizione in cui l'individuo possiede un cariotipo 46, XY e genitali esterni femminili. Nella maggioranza dei casi i soggetti hanno un'alterazione a carico di SRY.
Le donne con la sindrome di Swyer non hanno ovaie, né testicoli, ma delle gonadi "streak", cioè non funzionanti, per cui sono assenti sia le cellule germinali sia le cellule che producono ormoni. Vagina e utero sono normali (le così dette strutture Mulleriane).
Tuttavia, non essendo presenti gonadi funzionanti non sono prodotti gli ormoni necessari per lo sviluppo puberale e generalmente non si ha crescita del seno, mentre possono essere presenti peli pubici e ascellari. Alcune donne hanno il clitoride leggermente ingrandito La sindrome di Swyer viene spesso diagnosticata quando la ragazza viene visitata per un ritardo della pubertà; con la terapia ormonale si ottiene una maturazione dei caratteri sessuali secondari.
Come nel caso precedente la presenza di tessuto gonadico streak aumenta il rischio di tumori pertanto è necessaria la rimozione chirurgica.
Sindrome di Denys-Drash, sindrome di Fraser e sindrome WAGR: A causa di una mutazione a carico di WT1 (11p13) si verificano:
nefropatia con sindrome nefrosica a partire da 3 anni;
tumore di Wilms;
ambiguità dei genitali di vario grado con streaks, in particolare nei soggetti affetti che presentano cariotipo 46, XY.
Camptomelic syndrome: Alterazione a carico di SOX-9, gene su 17q24 preposto alla regolazione del collagene tipo II. Si associano:
incurvamento delle ossa lunghe;
malformazioni degli organi interni;
fenotipo femminile negli affetti 46, XY.
Deficit di 5α-reduttasi: Il deficit di steroido 5-alfa-reduttasi 2 è una rara malattia autosomica recessiva che causa pseudoermafroditismo maschile (MPH), una condizione caratterizzata da differenziazione incompleta dei genitali maschili in pazienti 46, XY. Vi sono aree al mondo in cui è singolarmente frequente (es. Repubblica Dominicana).
L'enzima 5-alfa-reduttasi 2, codificato dal gene SRD5A2, catalizza la conversione del testosterone (T) in diidrotestosterone (DHT), essenziale per la normale differenziazione dei genitali maschili esterni e lo sviluppo del seno urogenitale. Sono state descritte oltre 40 mutazioni in tutti e 5 gli esoni del gene SRD5A2, localizzato in 2p23. Si tratta soprattutto di sostituzioni aminoacidiche, ma anche di delezioni complete del gene, piccole delezioni, mutazioni nonsenso e mutazioni nei siti di splicing.
Il DHT è indispensabile per la maturazione dei genitali esterni e, in epoca puberale, per la maturazione della prostata e la crescita dei peli; per la mascolinizzazione dei dotti di Wolff basta il testosterone.
La sindrome classica (ipospadia pseudovaginale perineoscrotale) è caratterizzata alla nascita da genitali esterni ambigui, con fallo simil-clitoride, ipospadia, scroto bifido e persistenza del seno urogenitale, con sacco vaginale perineale a fondo cieco. Tuttavia, il fenotipo dei genitali esterni può variare tra quello di una femmina completa (tipo 5) al maschio con ipospadia e/o micropene (tipo 1). I testicoli sono localizzati nelle pieghe labioscrotali o nei canali inguinali. E' bene sviluppato il tratto urogenitale interno, mentre sono assenti le strutture che derivano dai dotti Mülleriani.
Durante la pubertà, ammesso che non sia stata eseguita la gonadectomia, si manifesta una virilizzazione significativa, senza ginecomastia, correlata all'azione del testosterone con crescita del pene, discesa dei testicoli, comparsa dei caratteri sessuali secondari e spermatogenesi (la maggior parte dei pazienti è comunque sterile).
Le indagini ormonali di base e dopo stimolazione con hCG (gonadrotropina corionica umana) rivelano livelli normali o elevati di testosterone, basso DHT e aumento del rapporto T/DHT (>20). La conversione di T in DHT può essere saggiata su colture di fibroblasti ottenuti dalla cute dei genitali, anche se sono frequenti i risultati falsi negativi.
L'attribuzione del sesso è ancora dibattuta e deve essere accuratamente discussa in ogni paziente, a seconda dei risultati attesi della chirurgia plastica tesa a mascolinizzare i genitali. Se viene attribuito il sesso femminile deve essere effettuata la chirurgia femminilizzante dei genitali e la gonadectomia. La diagnosi prenatale è possibile previa caratterizzazione delle mutazioni causali nei pazienti.
Pseudo-ermafroditismo femminile o virilizzazione: Può essere dovuto alla somministrazione alla madre gravida di ormoni virilizzanti quali androgeni o più frequentemente progestinici nei casi di minacce d'aborto; può raramente trattarsi di tumori materni secernenti ormoni quali androblastomi, luteomi o tumori di Krukenberg. La virilizzazione può poi avvenire per deficit dell'enzima aromatasi, che converte gli androgeni in estrogeni, o per iperplasia del surrene.
Sindrome adreno-genitale: Consiste nella modificazione istologica risultante dalla aumentata increzione cronica di ACTH e nelle alterazioni sistemiche dovute al deficit di produzione di cortisolo.
L'incremento di ACTH è causato da livelli bassi di cortisolo, la cui sintesi è compromessa dall'assenza o dalla riduzione di uno dei cinque enzimi necessari per la sua produzione dal colesterolo. Ciascun blocco enzimatico determina un deficit caratteristico e l'accumulo di precursori specifici degli ormoni surrenalici. Nelle forme più comuni di iperplasia surrenalica congenita (SAG), i precursori prossimali al blocco enzimatico si accumulano e sono convogliati verso la sintesi di androgeni surrenalici.
Quando il blocco enzimatico (p. es., il deficit di 21-idrossilasi) causa accumulo di androgeni, il disturbo che ne consegue è una forma virilizzante di SAG, causando gradi differenti di virilizzazione di un feto femmina affetto. Se il blocco enzimatico compromette la sintesi di androgeni, si ha una forma ipovirilizzante, per la virilizzazione inadeguata di un feto maschio affetto.
Diverse alterazioni autosomiche recessive possono essere causa di SAG. Un lattante affetto si può presentare con ambiguità dei genitali esterni, fornendo, quindi, pochi indizi diagnostici iniziali, perché un maschio ipovirilizzato e una femmina ipervirilizzata non possono essere distinti sulla base dell'esame obiettivo. L'esame dei genitali esterni rivela tipicamente una struttura simil-fallica, che sembra più lunga e più larga di un clitoride ma più piccola di un pene, un'apertura singola alla base di questo fallo, che è il seno urogenitale, e gradi diversi di fusione incompleta delle pieghe labio-scrotali.
Livelli basali di 17-idrossiprogesterone > 8 ng/ml sono praticamente diagnostici di SAG dovuta a deficit di 21-idrossilasi. È necessario il test di stimolazione con ACTH per distinguere le varie cause di SAG. Le concentrazioni dei precursori degli ormoni surrenalici sono determinate prima e 30 minuti dopo la somministrazione EV di 250 mg di ACTH sintetico. L'incremento e il rapporto dei vari precursori sono diagnostici di ciascun difetto enzimatico.
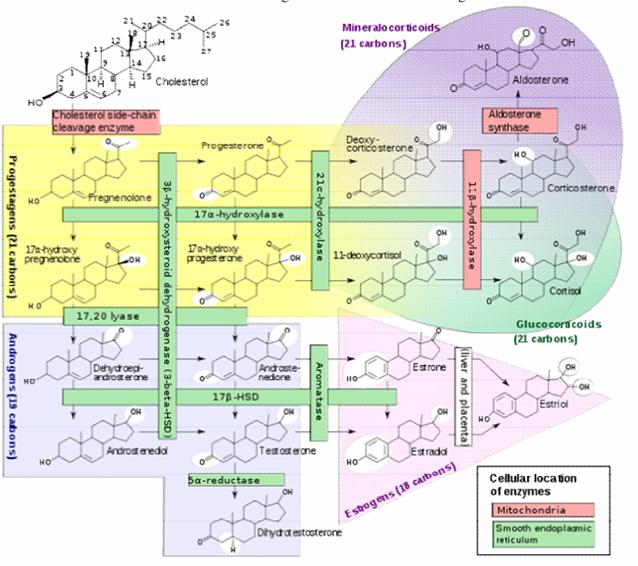
Figura : Steroidogenesi.
In alcuni dei deficit enzimatici meno gravi (p. es., deficit della 21-idrossilasi a esordio tardivo o della 11-b-idrossilasi), la virilizzazione può non diventare evidente che nella tarda infanzia, nell'adolescenza o durante l'età adulta. I sintomi possono comprendere ingrossamento del pene o del clitoride, irsutismo, seborrea, abbassamento del tono della voce, accelerazione dell'accrescimento con chiusura precoce delle epifisi (cartilagini di accrescimento delle ossa lunghe) che comporta bassa statura, aumento delle masse muscolari, calvizie in sede temporale, amenorrea e oligomenorrea durante l'età adulta.
Deficit di 21-idrossilasi: questo deficit causa il 90% di tutti i casi di SAG. L'incidenza oscilla da 1/10000 a 1/15000 nati vivi. C'è aumentata produzione di progesterone, 17-OH-progesterone, deidroepiandrosterone (DHEA), che è un androgeno debole responsabile di mascolinizzazione dei lattanti di sesso femminile affetti, e androstendione, con cortisolo e aldosterone plasmatici in basse concentrazioni o assenti. I metaboliti urinari di questi precursori (17-ketosteroidi e pregnantriolo) sono presenti in quantità superiori alla norma. La ridotta secrezione di aldosterone determina perdita di sale con iponatremia e iperkaliemia; l'attività reninica plasmatica è, pertanto, elevata. Nei difetti enzimatici parziali, il deficit di aldosterone non viene espresso e i pazienti sono normonatriemici e normokaliemici. Si è riscontrata un'associazione non casuale con alcuni aplotipi HLA. È possibile effettuare sia la diagnosi che il trattamento prenatali. Si può anche rilevare lo stato di portatore (eterozigote) nei bambini e negli adulti.
La terapia del deficit di 21-idrossilasi si effettua con la somministrazione sostitutiva di glucocorticoidi (idrocortisone, cortisone acetato o prednisone) e, quando necessario, con il ripristino della omeostasi normale di sodio e potassio con i mineralcorticoidi. La somministrazione orale di idrocortisone (da 15 a 25 mg/m2/die in 3 dosi) o prednisone (3-4 mg/m2 /die in 2 dosi) viene adeguata per mantenere i precursori degli androgeni entro l'intervallo appropriato per l'età. La terapia è finalizzata alla normalizzazione sia dell'androstendione, del 17-OH-progesterone e della attività reninica plasmatici che dei metaboliti urinari (17-ketosteroidi e pregnantriolo). Il fludrocortisone per via orale (0,1 mg/die) va somministrato se c'è perdita di sale. I lattanti spesso richiedono un supplemento di sale per os. È difficile il monitoraggio stretto durante la terapia. L'ipertrattamento con glucocorticoidi determina la malattia di Cushing iatrogena, che si manifesta nell'infanzia con obesità, crescita ridotta e ritardo dell'età ossea. L'ipo-trattamento con glucocorticoidi non riesce a sopprimere la secrezione di ACTH con conseguente iperandrogenismo, che si manifesta nell'infanzia con virilizzazione e velocità di crescita superiore alla norma e infine interruzione precoce dell'accrescimento con bassa statura finale.
Deficit di 11b-idrossilasi: questo deficit è responsabile del 3-5% di tutti i casi di SAG. Il profilo steroideo è caratterizzato dal l'incremento dell'11-deossicortisolo (e dei 17-idrossicorticosteroidi urinari) e del deossicorticosterone. A causa dell'attività mineralcorticoide del deossicorticosterone, i pazienti presentano ritenzione salina e ipertensione arteriosa con alkalosi ipokaliemica. L'attività reninica plasmatica è bassa. Si realizza anche virilizzazione. Il trattamento consiste nella terapia sostitutiva con cortisolo; può anche essere necessaria quella con mineralcorticoidi.
Deficit di 3b-idrossisteroidodeidrogenasi: questo disturbo molto raro comporta l'accumulo di DHEA, che viene convertito in testosterone perifericamente nel tessuto extrasurrenalico. Il trattamento consiste anche qui di glucocorticoidi con mineralcorticoidi quando necessario.
Deficit di colesterolo-desmolasi e di 17a-idrossilasi: questi disturbi provocano la virilizzazione dei lattanti femmine affetti e l'ipovirilizzazione di quelli maschi.
Deficit di corticosterone 18-metilossidasi di tipo II: la manifestazione è quella tipica della carenza di aldosterone: iperkaliemia cronica e bassa aldosteronemia. Non sono presenti alterazioni della differenziazione sessuale.
L'iperplasia congenita dei surreni
L'iperplasia congenita dei surreni (ISC) riguarda un gruppo di malattie associate a anomalie complete (forma classica) o parziali (non-classica) nella biosintesi degli ormoni del surrene. La prevalenza della forma classica associata al deficit della 21-idrossilasi è stimata in circa 1/14.000. Tuttavia, le forme non classiche sono comuni.
L'ISC è trasmessa come carattere autosomico recessivo. La forma più comune (95% dei casi) è causata dal deficit di 21-idrossilasi. Altre cause di ISC sono il deficit di 11-idrossilasi, 3-beta-idrossisteroido deidrogenasi, 17-alfa-idrossilasi.
La malattia è caratterizzata da un'insufficiente produzione di cortisolo o di aldosterone (forma classica con perdite di sali), associata a una iperproduzione di androgeni surrenali.
Nella forma classica, lo scompenso metabolico (disidratazione con iponatremia, iperkalemia e acidosi associata a deficit di mineralcorticoidi e ipoglicemia associata a deficit di glucocorticoidi) può costituire un rischio per la vita, a partire dal periodo neonatale. Nel neonato queste patologie possono portare alla morte se non riconosciute in tempo.
Alla nascita le femmine presentano genitali ambigui. L'iperadrogenismo può determinare un'accelerazione della crescita nell'infanzia e l'avanzamento dell'età ossea può esitare in una statura finale ridotta. Il bambino sembra che cresca bene però poi non crescono più e l'età ossea finale adulta risulta ridotta. Gli adulti presentano obesità, anomalie ossee e problemi di fertilità.
Le forme non classiche hanno un esordio tardivo nel periodo peri- o postpuberale e si manifestano con segni di iperandrogenismo (acne, irsutismo, problemi mestruali e infertilità).
Lo screening neonatale per il deficit della 21-idrossilasi (forma classica) è stato adottato in molti paesi e si basa sulla misurazione dei livelli di 17-idrossiprogesterone. (In italia non è ancora effettuato).
L'identificazione di un caso deve portare a indagare tutti i soggetti della famiglia e i consanguinei. La diagnosi prenatale è possibile con l'analisi molecolare del DNA fetale e permette di avviare la terapia per prevenire la virilizzazione nelle femmine affette.
La terapia ormonale sostitutiva (gluco- e mineral-corticoidi per la forma classica con perdita di sali, e glucocorticoidi per le forme semplici virilizzanti) va attuata per tutta la vita e richiede un attento follow-up (prima pediatrico, poi per l'adulto) e migliora la prognosi dei pazienti, in quanto previene le complicazioni associate all'iperandrogenismo cronico, permettendo di mantenere una normale fertilità. Le anomalie dei genitali nelle femmine possono richiedere interventi chirurgici.
Diagnosi di laboratorio
elevati livelli sierici di 17 beta estradiolo nella femmina e testosterone nel maschio;
bassi livelli di FSH e LH;
test al GnRH: risposta assente;
aumento DHEA-S;
aumento 17-OH progesterone.
Esami strumentali
età ossea avanzata;
ecografia pelvica: follicoli ovarici, eventuali masse pelviche.
Sindrome di Turner
Anomalia dei cromosomi sessuali in cui vi è una completa o parziale assenza di uno dei due cromosomi sessuali, con produzione di un fenotipo femminile. La sindrome di Turner si riscontra in circa 1/ 4000 femmine nate vive. Il 99% dei concepimenti 45, X esitano in aborti. L'80% delle nate vive con monosomia X ha perso il cromosoma X paterno. Le anomalie cromosomiche nelle femmine affette sono variabili. Circa il 50% ha un cariotipo 45, X.
Molte pazienti presentano mosaicismi (p. es., 45, X/46, XX oppure 45, X/47, XXX). Il fenotipo varia da quello tipico della sindrome di Turner a quello normale. In alcuni casi le donne colpite presentano un cromosoma X normale e un X che ha formato un cromosoma ad anello. In altri casi, invece, si riscontra un cromosoma X normale e un isocromosoma delle braccia lunghe, costituito da due braccia lunghe del cromosoma X per perdita delle braccia corte. Questi soggetti tendono ad avere molte delle caratteristiche fenotipiche della sindrome di Turner. In questo modo la delezione del braccio corto del cromosoma X sembra giocare un ruolo fondamentale nella determinazione del fenotipo della sindrome.
Clinica: le neonate colpite si possono presentare con marcato linfedema del dorso delle mani e dei piedi e con linfedema o pterigio della regione posteriore del collo. Comunque, molte femmine con sindrome di Turner sono colpite in maniera molto lieve. L'aspetto tipico è caratterizzato da bassa statura, pterigio del collo, bassa attaccatura dei capelli sulla nuca, ptosi palpebrale, torace largo con capezzoli molto distanziati, nevi pigmentati multipli, accorciamento del 4° metacarpo e metatarso, falangi distali prominenti con spirali nei dermatoglifi delle estremità delle dita, ipoplasia delle unghie, coartazione aortica, bicuspidia della valvola aortica e valgismo del gomito. Sono comuni malformazioni renali ed emangiomi. In alcuni casi possono comparire teleangectasie nel tratto gastrointestinale con conseguente emorragia digestiva. Raro è il ritardo mentale. Tuttavia in molti casi si riscontra una certa compromissione di una specifica abilità percettiva e di conseguenza un basso punteggio nei test attitudinali e matematici, anche se si riscontrano punteggi nella media e oltre nei test verbali relativi al calcolo del quoziente intellettivo. Nel 90% delle persone affette si riscontrano disgenesia gonadica con mancato sviluppo puberale, scarso sviluppo di tessuto mammario o assenza del menarca. La terapia sostitutiva con ormoni sessuali femminili consente lo sviluppo puberale. Le ovaie sono sostituite da banderelle di tessuto fibroso (streak), di solito prive di oocellule in via di sviluppo. Tuttavia, il 5-10% delle ragazze affette raggiunge il menarca spontaneamente, ma, molto raramente, sono risultate fertili e hanno avuto dei bambini.
Diagnosi: si deve effettuare un'analisi citogenetica e uno studio con sonde specifiche per il cromosoma Y in tutti i pazienti con disgenesia gonadica, per escludere casi di mosaicismo con una linea cellulare in cui è presente il cromosoma Y; p. es., 45,X/ 46,XY. Di solito questi soggetti presentano un fenotipo femminile con caratteristiche variabili di sindrome di Turner. Essi sono esposti ad alto rischio di tumori maligni delle gonadi, soprattutto gonadoblastoma e devono essere sottoposti ad asportazione profilattica delle gonadi, non appena effettuata la diagnosi.
Sindrome di Klinefelter
Anomalia dei cromosomi sessuali nella quale ci sono due o più cromosomi X e un Y, con conseguente sviluppo di un fenotipo maschile.
La sindrome di Klinefelter si osserva in circa 1/ 800 maschi nati vivi. L'extra cromosoma X è di origine materna nel 60% dei casi. Le persone affette tendono a essere alte, con braccia e gambe sproporzionatamente lunghe. Spesso hanno testicoli piccoli, atrofici e circa 1/3 sviluppa ginecomastia. La pubertà si presenta di solito all'età normale, ma spesso la crescita dei peli sul viso è scarsa. Si riscontra una certa difficoltà di apprendimento e in molti casi deficit specifici nel linguaggio, nel processo auditivo e nella lettura. La variabilità clinica è notevole e molti maschi 47, XXY sono normali per aspetto e intelligenza e vengono individuati nel corso di indagini effettuate per la sterilità (probabilmente tutti i maschi 47, XXY sono sterili) o di screening citogenetici eseguiti sulla popolazione normale. I maschi appartenenti a quest'ultimo gruppo sono stati seguiti nel corso dello sviluppo. L'incidenza dell'omosessualità non è aumentata. Lo sviluppo testicolare varia dalla presenza di tubuli ialinizzati e non funzionali, fino a una certa produzione di spermatozoi, mentre l'escrezione urinaria dell'ormone follicolo-stimolante è frequentemente aumentata.
Varianti: il mosaicismo si riscontra nel 15% dei casi. Alcuni individui affetti hanno 3, 4, o perfino 5 cromosomi X insieme al cromosoma Y. In generale con l'aumento del numero dei cromosomi X aumenta anche la gravità del ritardo mentale e delle malformazioni.
|
| Appunti su: sessualita27 nei maschi di 14 anni: puo27 essere ambigua3F, 21 alfa idrossilasi, bottoncino mammario femminile a 3 anni, pubertC3A0 precoce ecografia, 11 beta idrossilasi, |
|
| Appunti Nutrizione |  |
| Tesine Bellezza |  |
| Lezioni Bambini |  |