|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 1923 | Gradito: |
Leggi anche appunti:Distrofia muscolareDISTROFIA MUSCOLARE Chi ha scoperto la malattia e quando? La malattia Trapianto renaleTrapianto renale Il trapianto di rene è la terapia sostitutiva di scelta per i HelicobacterHELICOBACTER ♠ Bacillo , ricurvo |
 |
 |
EMOGLOBINOPATIE - BAMBINO
Tipi di emoglobina embrionale, fetale e adulta: due gruppi di geni sono coinvolti nella produzione di emoglobina che ricordiamo essere un tetramero composto da quattro catene globiniche, questi geni sono localizzati sul braccio corto del cromosoma 16 e del cromosoma 11.
Sul cromosoma 16 ci sono 3 geni all'interno del cluster del gene α: zeta (ζ), e due geni alfa (α1 e α2). Sul cromosoma 11 ci sono 5 geni all'interno del cluster del gene β: epsilon (ε), delta (δ), due geni gamma (γ) e beta (β). L'espressione di questi geni segue una tempistica ben precisa e dalla combinazione di essi si generano 6 tipi di emoglobine:

I rapporti tra i tipi di emoglobina dopo i 6-12 mesi sono sovrapponibili a quelle dell'adulto:
HbA (95%);
HbA2 (2-3,5%);
HbF(<2%).
Emoglobinopatie a cellule falciformi
Le emoglobinopatie a cellule falciformi comprendono:
L'anemia falciforme (omozigosi dell'emoglobina S o Drepanocitosi);
Le emoglobinopatie a cellule falciformi in cui all'eterozigosi per Hb S si associa una altra mutazione dell'altro allele β (Hb C, Hb D, Hb O, β talassemia).
Nell'anemia a cellule falciformi in genere la quantità di HbS è maggiore del 90%, nelle altre forme di emoglobinopatia è in genere >50%.
L'emoglobina S (Hb S) è il risultato del cambiamento di un singolo paio di basi (timina per adenina) al sesto codone del gene β globinico, che determina la sostituzione della glutammina con una valina in posizione sei della globina β,la sostituzione di un amminoacido neutro al posto di una carico determina:
Minore solubilità dell'Hb S in condizioni di bassa tensione di ossigeno;
Precipitazione e cristallizzazione dell'emoglobina all'interno del globulo rosso con formazione del tipico aspetto a falce;
Perdita di elasticità ed emolisi.
Tutte queste alterazioni a carico del globulo rosso determinano alterazioni emodinamiche del sangue con rallentamento del circolo ed aumento di fenomeni trombo-embolici.
Manifestazioni cliniche
Asplenia funzionale: tutti i bambini affetti da anemia a cellule falciformi presentano una minore protezione immunologica (probabilmente dovuta a ridotta attività della milza) verso batteri capsulati che li predispone a maggior rischio di infezioni o sepsi che possono anche essere mortali.
Febbre: la predisposizione verso le infezioni determina una storia clinica di febbri frequenti le quali in un bambino con anemia falciforme non devono essere mai sottovalutate ma subito trattate.
Sequestro spenico: soprattutto nei lattanti il sequestro splenico è una condizione che può mettere in pericolo la vita, esso consiste nella congestione improvvisa della milza che si ingrossa e trattiene sangue sottraendolo al circolo, si verifica quindi un ipovolemia ed anemia acuta (riduzione dell'emoglobina di 2 mg/dl rispetto ai valori di base). Questa condizione è un'urgenza medica che necessità di misure che mirino al ristabilite l'emodinamica tramite l'infusione di sangue o liquidi isotonici.Per prevenire le recidive è consigliata la splenectomia.
I fenomeni vaso-occlusivi: L'ischemia del microcircolo causa ripetuti episodi di dolore in questi bambini che devono essere trattati con paracetamolo o addirittura con codeina e oppioidi, quando tali fenomeni occlusivi si verificano a livello celebrale si possono verificare vere e proprie ischemie con alterazioni neurologiche della durata superiore alle 24 ore e alterazioni all'RM che mostrano le aree ischemiche.
Priapismo: Erezione dolorosa della durata maggiore di 30 minuti (il trattamento prevede l'uso di farmaci per il dolore e se l'erezione non si risolve per più di 4 ore si ricorre all'aspirazione di sangue dai corpi cavernosi seguita da iniezione di adrenalina).
Sindrome toracica acuta: consiste in un insieme di reperti che includono:
Una nuova opacità radiografica;
Febbre;
Distress respiratorio;
Dolore toracico.
Non è ancora chiaro se tale sindrome sia causata da fenomeni vascolari o sia correlabile a fenomeni infettivi. La terapia si basa su procedure che mirino alla profilassi delle citate manifestazioni cliniche:
Per prevenire le infezioni è indicata la profilassi antibiotica con penicillina V;
Della terapia del dolore già abbiamo parlato (tachipirina, codeina o oppioidi), l'idrossiuurea, un agente mielosoppressore sembra aver dato buoni risultati nel ridurre gli episodi dolorosi (il meccanismo probabilmente riguarda l'innalzamento dei livelli di Hb F);
Le exanguinotrasfusioni (prelevare una certa quantità di sangue e sostituirlo con la stessa quantità di sangue sano) permettono di abbassare la percentuale di Hb S nel sangue e ciò riduce il rischio di complicanze, tuttavia l'accumulo di ferro è molto frequente nonostante l'uso di agenti chelanti.
Sindromi talassemiche
Le sindromi talassemiche sono delle emoglobinopatie ereditarie trasmesse con carattere autosomico recessivo, caratterizzate da ridotta o assente sintesi di una o più catene peptidiche costituenti l'emoglobina. Si dividono in:
α talassemie:il deficit riguarda le catene α;
β talassemie: il deficit riguarda le catene β.
In tutte le forme di talassemia l'alterazione dei normali rapporti di produzione tra le varie catene globiniche determina la formazione di emoglobine anormali questo determina:
compromissione della normale maturazione dei globuli rossi nel midollo; si stabilisce quindi un quadro di eritropoiesi inefficace con un iperattività del midollo ma morte prematura delle cellule (pochi reticolociti);
Precipitazione delle emoglobine anomale ed emolisi.
Tutto questo determina un quadro di anemia microcitica ipocromica con globuli rossi di morfologia alterata (possono ritrovarsi ad esempio le tipiche cellule a bersaglio).
α talassemia
Quando il difetto di produzione riguarda le catene α vi è un eccesso di catene β e γ, ciò determina durante la vita fetale (espressione maggiore di catene γ) la formazione della cosiddetta emoglobina di Bart (γ4), e dopo la nascita l'emoglobina H (β4).
Le catene α sono prodotte dai geni α globinici (4 geni) localizzati sul cromosoma 16 (due geni per ogni cromosoma), i quadri ematologici saranno più o meno gravi a seconda che il difetto coinvolga uno o più geni:
Delezione di 1 solo gene: Portatore silente (valori ematologici normali o lieve anemia microcitica ipocromica);
Delezione di 2 geni: Portatore di alfa talassemia (anemia microcitica);
Delezione di 3 geni: Malattia da Hb H (anemia microcitica, epatosplenomegalia, anomalie scheletriche);
Delezione di 4 geni: Idrope fetale (non compatibile con la vita).
β talassemia
Nelle β talassemie è presente un eccesso di catene in confronto alle γ e alle α, si formano quindi tetrameri (α4), queste inclusioni interagiscono con la membrana del globulo rosso e riducono la sopravvivenza eritrocitaria. Le catene sono prodotte in crescente quantità cosi come si verifica un aumento dell'emoglobina fetale (Hb F) e dell'HbA2.
Il gene mutato può determinare un'assente produzione delle catene β (β°) o una produzione ridotta (β+) con quadri clinici estremamente variabili:
Condizione di eterozigosi: Tratto talassemico clinicamente silente (portatore sano);
Condizione di omozigosi:
Talassemia major o morbo di Cooley: esordio dopo il 6° mese di vita, quando l'Hb F viene quasi completamente sostituita da HbA, il quadro clinico comprende grave anemia microcitica ipocromica ed epatosplenomegalia;
Talassemie intermedie si verificano per:
omozigosi per forme lievi;
coesistenza di α talassemia;
persistenza genetica di Hb F.
Sono tutte condizioni in grado di bilanciare l'eccesso di catene α e che quindi determinano un quadro clinico meno grave.
Diagnosi delle talassemie: La diagnosi di certezza viene effettuata mediante elettroforesi dell'emoglobina che mostra l'alterazione del normale rapporto tra le emoglobine e la presenza di emoglobine anomale.
Terapia delle talassemie
Trasfusioni (in genere mensili) più deferoxamina (chelante il ferro) per posticipare l'emosiderosi trasfusionale;
Splenectomia per ridurre l'emolisi;
Trapianto di midollo (forme gravi).
Anemie emolitiche autoimmuni
Le anemie emolitiche autoimmuni possono essere causati da una serie di agenti esterni e patologie, una variante emolitica è rappresentata dall'anemia emolitica isoimmune causata dal trasferimento transplacentare di anticorpi materni diretti contro i globuli rossi del feto.
Indipendentemente dalla causa che ne ha determinato la produzione questi anticorpi opsonizzano la membrana del globulo rosso e ne determinano l'emolisi o il sequestro ad opera del sistema reticolo-endoteliale.
Si distinguono:
Anemie emolitiche da anticorpi caldi (sono in genere IgG attivi tra i 35°-40°, e non richiedono il complemento per l'attività);
Anemie emolitiche da anticorpi freddi (attivi a tempreture più basse, attivano il complemento);
Anemia emolitica autoimmune da anticorpi caldi:
Primaria (idiopatica);
Secondaria a: patologie linfoproliferative, Lupus eritematosi sistemico, Malattie infiammatorie croniche, Artrite reumatoide, Infezioni virali (Epsein Barr, citomegalovirus ecc.), Immunodeficienze, Farmaci (penicilline, cefalosporine);
Anemia emolitica autoimmune da anticorpi freddi:
Primaria (idiopatica);
Secondaria a: patologie linfoproliferativo, infezioni (micoplasma pneumoniae, epstein barr virus).
La diagnosi di anemia autoimmune si basa sul riscontro del test di Coombs diretto o indiretto positivo (il test diretto evidenza la presenza di globuli rossi opsonizzati da autoanticorpi, il test indiretto evidenzia invece la presenza di anticorpi liberi nel siero).
Spesso le anemie autoimmuni sono autolimitanti e non necessitano di trattamento,quando però compare una porpora trombocitopenia concomitante (sindrome di Evans) la prognosi può essere spesso infausta.
Il trattamento nelle forme di anemia grave si basa sulla somministrazione di glucocorticoidi (il meccanismo d'azione si basa sulla riduzione dell'attività dei macrofagi del sistema reticolo-endoteliale e la riduzione della produzione anticorpale).
Possibile iter diagnostico
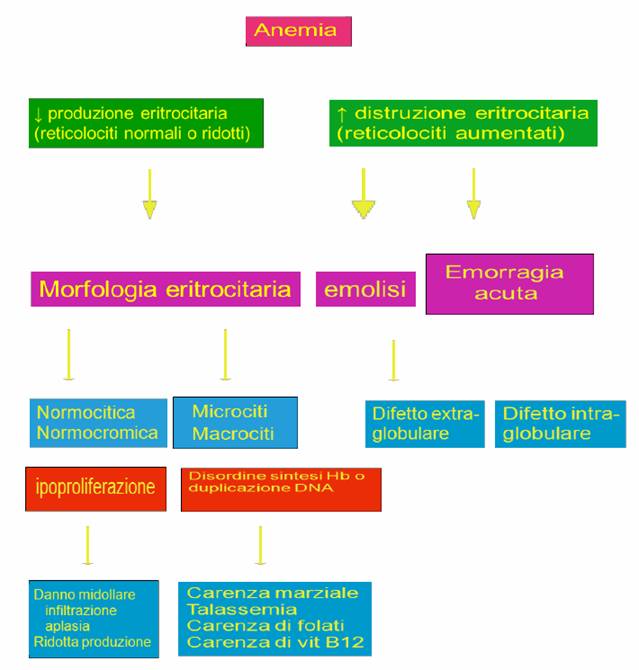
|
| Appunti su: |
|
| Appunti Nutrizione |  |
| Tesine Bellezza |  |
| Lezioni Bambini |  |