|
| Appunti universita |
|
|
| Appunti universita |
|
| Visite: 2819 | Gradito: |
Leggi anche appunti:Adenocarcinoma polimorfo di basso gradoAdenocarcinoma polimorfo di basso grado Si decide IgieneIgiene Siamo sempre nel campo della prevenzione primaria della malattia Il neonato di madre tossicodipendenteIl neonato di madre tossicodipendente 1 Come identificarlo La diffusione |
 |
 |
DEMENZE NEURODEGENERATIVE: DALLE BASI BIOMOLECOLARI AI PROTOCOLLI DIAGNOSTICI E TERAPEUTICI
La demenza è un concetto clinico e riguarda la perdita delle funzioni cognitive.
Noi parleremo delle D. neurodegenerative, che sono la gran parte di quelle conosciute, e di quelle vascolari come uniche D. non neurodegenerative.
Le D. possono essere classificate in senso generale attraverso vari tipi di paradigmi:
CLASSIFICAZIONE IN BASE
ALL'ETA' DI ESORDIO 1. Demenze Presenili
2. Demenze Senili
(considerando come cut-off l'età di 65 anni)
CLASSIFICAZIONE IN BASE ALLA SEDE DELLE LESIONI
1. Demenze Corticali:
· D. Di
Alzheimer
· D. Con
Corpi Di Lewy
· D.
Fronto-temporale (Malattia Di Pick)
2. Demenze Sottocorticali:
· D.
Vascolari
· D. Con
Disturbi Del Movimento
Corea
Di Huntington,
Morbo
Di Parkinson,
Paralisi
Sovranucleare Progressiva,
Degenerazione
Cortico-basale
Bisogna ricordare che nella stragrande maggioranza dei casi:
![]()
![]() D. corticali degenerative
D. corticali degenerative
D. sottocorticali possono non essere degenerative, ma vascolari o da idrocefalo normoteso(e di solito associate a disturbi motori cioè sia di foza, sia di continuità che di organizzazione di un movimento)
CLASSIFICAZIONE IN BASE ALL'EZIOLOGIA
Questa è una classificazione estremamente importante soprattutto ai fini clinici
Demenze primarie o primitive:(sono D. che non hanno una grande capacità di essere revertite per macanza di una terapia eziopatogenetica adeguata)
D. di Alzheimer
Malattia di Pick
D. da prioni
D. da malattie degenerative del S.N.C.(Morbo di Parkinson, corea di Huntington, paralisi sopranucleare progressiva, epilessia Mioclonica progressiva)
Demenze secondarie:( sono potenzialmente reversibili)
· D. vascolari ( D. Multi-infartuale, D. Da singoli infarti, D. Da coinvolgimento di piccoli vasi, D. Da ipoperfusione, D. Da emorragia)
· D. endocrine e metaboliche
· D. carenziali (D. Alcolica, sindrome Wernicke-Korsakoff, pellagra, deficienza di vitamina B12 e Folati)
· D. da agenti tossici (metalli pesanti, monossido di carbonio, piombo, arsenico, manganese, alluminio, farmaci -sono quelle che provochiamo noi medici nei soggetti con multi patologie che hanno bisogno di associare più farmaci!-)
· D. da neoplasie cerebrali (tumori Intracranici)
· D. da idrocefalo normoteso (può provocare una demenza sottocorticale progressiva potenzialmente reversibile)
· D. da traumi cranici (ematomi, emorragie, Ipossia, encefalopatia cronica dei pugili)
· D. da infezioni (infezioni HIV, meningiti croniche, encefaliti, sclerosi multipla)
CLASSIFICAZIONE IN BASE ALLA PROGNOSI
Demenze Degenerative o irreversibili perché sono degenerative e contro la neurodegenerazione non abbiamo terapia:
· D. di Alzheimer
· D. Neurodegenerative di tipo non Alzheimer
D. Fronto-Temporale
Malattia di Pick
D. con Corpi di Lewy
Malattia di Parkinson
Sindromi Parkinsoniane Atipiche
Corea di Huntigton
Degenerazione Cortico-Basale
Paralisi Sopranucleare Progressiva
Demenze Non Degenerative o Reversibili (sono una discreta parte delle Demenze Secondarie e costituiscono meno del 15% di tutte le Demenze):
· D. da Idrocefalo Normoteso
· D. da Cause Infettive
· D. da Cause Metaboliche
· D. da Lesioni Occupanti Spazio
· D. da Cause Psichiatriche (PSEUDODEMENZA)
EPIDEMIOLOGIA
Dal punto di vista epidemiologico, nel mondo occidentale abbiamo una sostanziale prevalenza della malattia di Alzheimer ,che ragginge il 60-70% di tutte la demenze, mentre nel mondo asiatico la patogenesi più frequente delle demenze è quella vascolare.
Questo tipo di andamento epidemiologico è continuamente sovvertito dal progresso delle conoscenze,infatti fino a 50 anni fa quasi tutti i disturbi cognitivi si consideravano vascolari( aterosclerotici);pochi decenni dopo tali disturbi cominciarono ad attribuirsi tutti all'Alzheimer e fino a 20 anni fa di demenze non-Alzheimer se ne conoscevano ancora poche.
Un concetto importante è che l'incidenza e la prevalenza della demenza, qualunque essa sia,aumentano con l'età. In base a questo ragionamento a 90 anni 1 su 3 sarà demente e siccome abbiamo detto che la maggior parte delle demenze sono di tipo Alzheimer, si potrebbe pensare che l'Alzheimer sia solo espressione del fisiologico invecchiamento,cioè, se noi vivessimo abbastanza dovremmo diventare tutti dementi!Per alcuni studiosi è così ma per molti,compreso io, non lo è. Sulla base di studi epidemiologici, infatti, si è visto che la curva dell'incidenza non cresce esponenzialmente con l'età bensì diventa asintotica, cioè si appiattisce. Questo vuol dire che l'Alzheimer è una malattia e non un'espressione dell'invecchiamento cerebrale.
La demenza è una condizione altamente invalidante e in continuo aumento in virtù dell'invecchiamento della popolazione. È diventata una malattia a forte impatto sociale non solo per le sue dimensioni, ma soprattutto perché è mutato il micro-ambiente nel quale è gestita: ormai è fortemente diminuito il numero di quelle donne casalinghe che un tempo potevano prendersi cura di chi aveva sviluppato una demenza. Così oggi queste persone affette sono in continua ricerca di un'occupazione o di un luogo di ricovero e il costo per l'assistenza di questi pz è estremamente aumentato.
DEMENZA VASCOLARE
Prima di passare alle D. neurodegenerative, facciamo un accenno alla demenza vascolare perché riveste una notevole importanza.
Costituisce circa il 10-15% di tutte le demenze.
Sono stati proposti criteri per la diagnosi di demenza vascolare probabile e che ci permettono anche di distinguerla da quella degenerativa:
evidenza clinica di demenza
evidenze cliniche e di neuroimaging (TAC, RMN) di malattia cerebrovascolare
relazione evidente o indiretta tra la demenza e la malattia cerebrovascolare (esordio, fluttuazioni, deterioramento 'a scalini' dei deficit cognitivi).
Un elemento fondamentale è la progressione della malattia che, essendo vascolare, ha un andamento non progressivo ma "a gradini", per l'aumento del carico lesionale o per l'insorgenza di un nuovo infarto in una posizione strategica
sottotipi
Demenzamulti-infartuale(MID).
E' la risultante di infarti multipli e completi, generalmente nel territorio di
distribuzione, corticale o sottocorticale, dei grossi vasi. Questo provoca la
perdita di sostanza cerebrale e la conseguente interruzione della continuità
tra i circuiti neuronali: maggiore è la compromissione vascolare, cioè il numero
degli infarti, maggiore sarà il grado di demenza.
Demenza da singoli
infarti strategici.
E' data dai singoli infarti in aree cerebrali funzionalmente importanti per le
prestazioni cognitive (es. area mediale del lobo temporale dove risiedono le
strutture coinvolte nella memoria; giro angolare; proencefalo basale; talamo,
che è un nucleo a proiezione diffusa; silviana di sn il cui infarto dà una
grande compromissione parieto-temporale cortico-sottocorticale di sn e chi ne è
colpito sarà un grande afasico; etc.).
Demenza da coinvolgimento
dei piccoli vasi.
E' l'esito di lesioni ischemiche a carico dei vasi di piccolo calibro che
irrorano le strutture sottocorticali.
Demenza da ipoperfusione.
E' la risultante di un danno ipossico acuto, cronico o ripetuto.
Demenza emorragica.
E' la sequela di lesioni emorragiche intraparenchimali (fra le più frequenti
l'emorragia intracerebrale a sede capsulare) o extraparenchimali (ematoma
subdurale cronico, emorragia subaracnoidea).
Di fronte a tutti questi quadri dobbiamo considerare la possibilità che si sovrappongano più processi: per es. al danno vascolare discretamente importante di un soggetto iperteso si aggiungerà l' impoverimento neuronale parafisiologico che avviene con l'invecchiamento provocando così un deterioramento progressivo delle funzioni cognitive. In questo caso l'invecchiamento ha aggravato un danno di fondo e la demenza che ne deriva viene detta mista (cioè neurodegenerativa e vascolare insieme).
M. Di Alzheimer
Epidemiologia
È la più frequente demenza nel mondo occidentale
(dal 50% al 80% dei casi di demenza)
Può esordire nel presenio (prima dei 65 anni) , ma l'incidenza aumenta con l'aumentare dell'età
In più del 90% dei casi si presenta in forma sporadica.
Le forme familiari (5-7%) sono legate a mutazioni di geni differenti, quasi sempre capaci di determinare iperproduzione di b-amiloide
L'incidenza della malattia aumenta con l'età fino ad un certo punto, poi comincia a decrescere, il che è classico di tutte le malattie nosograficamente identificate e che hanno eziologia e patogenesi definite. Se la malattia fosse correlata all'età avremmo avuto un aumento esponenziale dell'incidenza ma c'è da dire che ciò non avviene anche per un errore che ci si porta dietro in questi studi, e cioè che sono poche le persone che arrivano oltre una certa età.
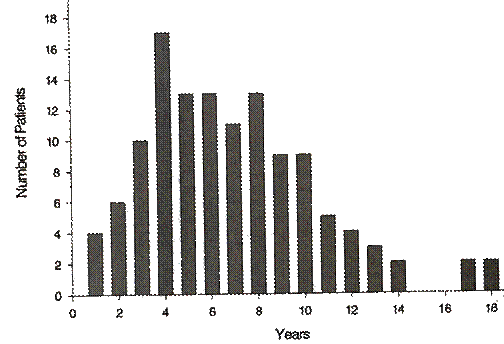
Questa è una proiezione un po' severa che risale a prima della terapia, quando effettivamente a 6 anni circa dall'esordio della malattia la demenza era già grave.
Esistono altri dati epidemiologici che sono significativi, in particolare quella che è la correlazione della malattia con uno specifico tipo di apolipoproteina: l'Apo-E.
Sappiamo che come tutte la apolipoproteine anche questa contribuisce al trasporto del colesterolo nei tessuti ma tra tutte è quella che più agevolmente lo porta nel tessuto nervoso centrale dove il colesterolo è un costituente fondamentale della mielina e delle membrane neuronali.
Si è visto che c'è una maggiore tendenza all'Alzheimer nei soggetti che hanno una Apo-E4 , laddove i fenotipi possibili sono E2, E3, E4 e ognuno di noi può avere una qualunque combinazione tra questi alleli: E2-E2, E3-E3, E4-E4,E2-E3,E3-E4,ecc. Già con un solo allele E4 il rischio di Alz. è aumentato e lo è molto di più quando ce ne sono due e tale rischio è correlato con la forma late-onset, cioè ad esordio tardivo.
Aspetti clinici
L'A. si presenta spt con un disturbo della memoria (il pattern è ipomnestico) e ciò ha un suo correlato anatomico dal momento che la malattia inizia nelle strutture mediali del lobo temporale, in particolare nell'ippocampo(disturbi mnesici). Da qui si diffonde alla corteccia entorinale, alla corteccia temporale in toto, in alto verso la corteccia parietale (disturbi del linguaggio) , per poi diramarsi alle aree posteriori (occipitali->disturbi visuo-spaziali) e anteriori (frontali->così spesso si associano anche disturbi del comportamento che consistono per es. nell'esasperazione di difetti caratteriali precedenti la malattia come la gelosia che può arrivare ad essere un delirio). L'Alzheimeriano si rende conto del suo deterioramento cognitivo e può sviluppare una depressione.
Prima fase
- Lieve perdita della memoria per gli eventi recenti, dimentica gli impegni
- Il pz diventa ripetitivo
- Tende a perdersi in ambienti nuovi
Inizia ad essere disorientato nel tempo
Il pensiero astratto risulta impoverito, con ridotta capacità di ragionamento logico e di giudizio
Progressiva incapacità a svolgere compiti prima semplici
Spesso vengono esagerati i caratteri premorbosi della personalità
Fase intermedia:cominciano i disturbi della sfera del linguaggio a partire da un "pensiero interciso"(cioè mentre parla si interrompe perché dimentica ciò che stava dicendo), per arrivare ad una "afasia nominum"(non sa nominare gli oggetti o le persone pur riconoscendoli dunque utilizza delle circonlocuzioni)
Il pz diviene incapace di apprendere nuove informazioni
Si perde spesso in ambienti a lui familiari
La memoria remota è compromessa
In genere completo disorientamento temporo-spaziale (cominciano a perdere anche la giusta sequenza temporale del proprio sé: "trasposizione diacronica", cioè sovrappongono diversi momenti della loro vita e magari si convincono di avere un figlio di dieci anni più piccolo,di avere ancora il genitore defunto in vita, ecc)
Richiede una certa assistenza nelle attività della vita quotidiana
Ulteriore compromissione del comportamento
Fase avanzata
Il pz è incapace a svolgere qualsiasi attività della vota quotidiana
Memoria a breve e lungo termine completamente persa
evoluzione verso il mutacismo e l'acinesia
Il pz diviene incapace di riconoscere anche i propri familiari
Incontinenza sfinterica(non per compromissione piramidale, ma perché non riconoscono più il significato dello stimolo che avvertono)
Disfagia(quando arrivano ad essere compromesse anche le strutture sottocorticali)
Diagnosi
Anche per l'A. la sola diagnosi di certezza è quella neuropatologica e in vivo il massimo grado diagnostico che si può raggiungere è quello di probabilità. Per farla dobbiamo avvalerci di:
Anamnesi :per percorrere la storia della malattia che sarà stata subdola
Analisi di laboratorio: dovranno escludere la presenza di malattie potenzialmente curabili (metaboliche,luetiche, idrocefalo normoteso, ecc)
ENPS: valutazione neuropsicologica che serve per visualizzare il profilo del deterioramento cognitivo del pz e quindi di risalire al tipo di strutture anatomiche coinvolte.
RMN (studi morfologici e volumetrici)ci permette di scoprire un eventuale tumore o problemi vascolari importanti che abbiano provocato la demenza.
All' RMN è possibile fare una diagnosi differenziale tra Alzheimer e non-Alzheimer :
1)misurando le dimensioni del corno temporale del ventricolo laterale che nell'A. si dilata in seguito all'atrofia del parenchima
2)tracciando il contorno dell'ippocampo (che nell'A. è diminuito di volume)e confrontandolo con il profilo di una persona sana per valutarne il deficit volumetrico.
SPECT/PET sono indagini funzionali;
Nell'A. l'ipoperfusione sarà spt temporale e parietale: il colore rosso è quello di un'area con un'attività metabolica normale: quando non c'è vuol dire che ci troviano di fronte a deficit di perfusione o di metabolismo.
Rachicentesi (dosaggio tau e -amiloide) Attualmente la ricerca è volta alla scoperta di marcatori biologici che ci permettano di diagnosticare quanto più precocemente possibile la malattia o addirittura di individuare i pz che la svilupperanno nel corso degli anni! Tutto ciò non è ancora possibile ma durante la malattia qualche informazione ci può essere data dalla proteina TAU e dalla B-amiloide; in particolare si valuta il "movimento" relativo di una rispetto all'altra che ci dà un elemento di diagnosi differenziale con altri tipi di demenze: maggiori sono le variazioni di queste proteine e più è probabile che si tratti di Alzheimer.
Esclusione di qualsiasi altra demenza secondaria
Alla biopsia notiamo una distribuzione diffusa dell'atrofia a differenza di quanto accade in altre demenze con atrofia focale.

Questa immagine rappresenta lo "spreading pattern" dell'A.
La malattia parte dalla corteccia entorinale, si dirama all'ippocampo,poi sale su allargandosi a tutto il lobo temporale per arrivare in seguito alla corteccia parietale, frontale e occipitale. Allo stesso modo si comporta il deficit dal punto di vista clinico.
Tutto ciò è stato dimostrato non solo con lo studio cerebrale ma anche in risonanza magnetica.
Basi Neuropatologiche
Esistono dei markers (imprescindibili)che ci permettono di fare diagnosi di A.:
1)PLACCHE SENILI
Depositi extracellulari amorfi, compatti contenenti nella parte centrale fibrille di b-amiloide, peptide (digerito male)derivante da una proteina denominata APP (Amyloid Precursor Protein) circondata da processi gliali, cellule microgliali e neuriti rigonfi in via di degenerazione.
La deposizione di queste placche si realizza non solo nel neuropilo, cioè a livello della corteccia, ma anche nelle strutture delle arteriole e delle venule cerebrali.
APP (Amyloid Precursor Protein)
- è una proteina integrale di membrana di 697-770 aminoacidi ubiquitaria
- è codificata da un gene localizzato sul cromosoma 21
- lo splycing alternativo del gene può dare 3 proteine di differente lunghezza: 695, 751, 777.
Normalmente la proteina viene tagliata nel sito dell'a-secretasi, che ne forma 2 frammenti che vengono normalmente metabolizzati dalle cellule. Fisiologicamente il 5% di questa proteina viene tagliato anche dalla β- e γ-secretasi che ne formano un frammento, detto Aβ, in quantità tali da non farlo accumulare. Nell'A. l'attività della β e γ-secretasi è aumentata così il frammento che ne deriva è in eccesso e si accumula; questa, che fino a poco fa era solo un'ipotesi, è stata confermata dal riscontro di famiglie in cui l'A. si trasmette geneticamente perché portatrici di mutazioni della APP che ne riducono l'affinità per l'a-secretasi e ne aumentano quella per la β e γ-secretasi.
- la forma di APP-695 è espressa ad elevati livelli solo nel SNC
-
la b-amiloide
è un peptide di lunghezza variabile da
Non si conosce la funzione dell'APP ma sembra che abbia un ruolo nella protezione della cellula contro l'eccitotossicità
Non esistono solo mutazioni del gene del precursore della β-amiloide, ma anche delle mutazioni su altri cromosomi e su altri geni possono dare A.:
Nella Trisomia del 21il gene APP viene a trovarsi in quella parte del cromosoma 21 sovrannumerario che trasloca nel gamete con trisomia; questo provoca una sovrapproduzione della β-proteina e spiega perché TUTTI i Down si ammalano di Alzheimer precocemente.
Altri 2 geni importanti sono la presenilina 1 e 2 (PS1e PS2)che partecipano alla regolazione dell'attività della γ-secretasi che risulta aumentata in caso di mutazioni di PS1 ePS2.
Infine
ci sono geni di ultima generazione come
Tutte queste mutazioni hanno in comune la capacità di indurre un aumento della produzione di β-amiloide.
N.B. L'accumulo di β-amiloide non sempre si correla con la neurodegenerazione (e quindi con la clinica), infatti il cervello di un anziano non demente può essere disseminato di placche quanto quello di un alzheimeriano pur senza presentare demenza. E'la neurodegenerazione il vero fattore che si correla con la clinica dell'A.
Un altro elemento, che è molto più correlato con la clinica è la:
2)DEGENERAZIONE NEUROFIBRILLARE
Fascicoli di filamenti appaiati ad elica, intraneuronali, composti da proteina Tau prevalentemente iperfosforilata
Il citoscheletro dei neuroni risulta destrutturato,disorganizzato, proprio perché la proteina TAU ha perso la sua corretta funzione cioè quella di MAP protein(proteina associata ai microtubuli).Le MAP assicurano il mantenimento, l'assemblaggio, il corretto di stanziamento, ecc. dei microtubuli perché, grazie ad un'attività di fosfo- e defosforilazione ,permettono ai microtubuli di allungarsi e accorciarsi all'occorrenza. Non dimentichiamo che i microtubuli sono strutture fondamentali per trasporto assoplasmatico e quindi per il funzionamento e la sopravvivenza dei neuroni.
La proteina TAU: è una proteina cellulare che assembla la tubulina in microtubuli e stabilizza i microtubuli nella struttura portante del citoscheletro.
Nella malattia di A., non sappiamo perché, ma questa proteina si iperfosforila e proprio quando mutata e/o iperfosforilata perde parte della sua capacità e si accumula nel citoplasma non solo dando origine alla degenerazione neurofibrillare ma anche partecipando alla formazione dei corpi di Pick, corpi di Lewy, degenerazione granulovacuolare, etc.
Tau fa parte del patrimonio proteico dei neuroni e della glia, ma non tutti i neuroni né tutta la glia degenerano se tau è malata.
Le tauopatie sono malattie prevalentemente focali che esitano in atrofie lobari o di porzioni di lobo cerebrale e/o del sistema extrapiramidale
Si conoscono 25 mutazioni del gene Tau che segregano con tauopatie familiari
Ricapitolando:
I markers neuropatologici fondamentali dell'A. sono 2:
Deposizione di β-amiloide (si realizza attraverso la formazione di placche senili)
Degenerazione neurofibrillare (dovuta al mal funzionamento della proteina TAU che accumulandosi all'interno della cellula, porta alla formazione di "tangles" che sono tossici per la cellula e provocano la destrutturazione del citoscheletro)
Sembra che l'accumulo di β-amiloide dia avvio alla trasduzione di un segnale che indurrebbe la formazione di tangles, stabilendo così una correlazione tra questi due eventi.
Terapia
Per la malattia di A. non c'è una terapia!
°Attualmente sono disponibili dei farmaci ( Donepezil, Rilastigmina e Galantamina) che non intervengono sul decorso della malattia ma sui sintomi: si tratta di anticolinesterasici centrali che agiscono sui recettori muscarinici(N.B. si tratta dei recettori colinergici CENTRALI, quelli periferici sono detti nicotinici) aumentando il tono colinergico. Il razionale di questa terapia si fonda sull'evidenza che nelle fasi precoci della malattia c'è una degenerazione notevole del nucleo basale del Meinert e di altri nuclei colinergici a proiezione diffusa (ma spt temporale e frontale)e di conseguenza una riduzione del tono colinergico. Le cellule a valle si trovano a non ricevere abbastanza acetilcolina, che noi aumentiamo con questi farmaci migliorando spt i disturbi cognitivi. Nonostante a lungo andare questa terapia non sia più efficace e dia effetti collaterali come nausea e vomito, va fatta sempre nei pz con A.
°Un
altro farmaco utilizzato è
°Esistono infine dei farmaci in via di sperimentazione e che per questo vengono somministrati solo previo consenso infomato:
Bracker della β-amiloide che entrano nella placca e la distruggono.
Farmaci contro l'iperfosforilazione della proteina TAU come i Sali di litio che venivano utilizzati contro la psicosi maniaco depressiva.
Sul piano internazionale fecero grande scalpore i vaccini preparati con proteine screb di β-amiloide: somministrati nei topi li proteggevano dallo sviluppo delle placche, ma negli uomini provocarono encefaliti mortali!(per effetto della cross-reazione tra gli antigeni del vaccino e la β-proteina self del cervello umano)
DOMANDA: è mai capitato che un soggetto rispondesse a tutti i criteri clinici di A. ma poi la diagnosi è stata smentita al rilievo autoptico?
Certo che succede!!!
Taupatie
La proteina Tau, da sola, può essere causa di altre malattie degenerative complessivamente definite come taupatie ;sono tutte a distribuzione lobare e il cui elemento patogenetico è la degenerazione neurofibrillare.
Degenerazione fronto-temporale
Demenza frontale
Afasia non fluente progressiva
Demenza semantica
Degenerazione corticobasale
Paralisi sopranucleare progressiva
Gliosi sottocorticale progressiva
Agnosia visiva progressiva
Aprassia progressiva
Compromissione progressiva dell'esplorazione spaziale
Astereognosia progressiva

Demenza fronto-temporale
Quello delle demenze fronto-temporali è il capitolo più importante, perché più corposo, sulle taupatie. In realtà con questa denominazione si intende un"complesso"di malattie neurologiche distinte.
La demenza fronto-temporale propriamente detta coinvolge primitivamente le strutture prefrontali e temporali ma esistono soggetti con un coinvolgimento prevalentemente prefrontale che avranno disturbi del comportamento e saranno apatici, abulici, inerti, che non si importano neanche di un parente che sta male, o disinibiti moriatici(a seconda delle porzioni della corteccia frontale coinvolte), e soggetti con un coinvolgimento prevalentemente temporale che saranno disfasici piuttosto che colpiti da disturbi comportamentali.
È responsabile del 20% dei casi di demenza. Prevale nell'età media (45-65aa).
Sul piano neuropatologico: atrofia fronto-temporale con perdita neuronale e gliosi che investe anche la sostanza bianca
Nella tabella che segue sono indicate la caratteristiche cliniche dominanti, a seconda dell'area prevalentemente colpita
Corteccia prefrontale dorsolaterale: soggetti con perseverazione, difficoltà di giudizio, disturbi nella pianificazione, ecc.
Corteccia orbito frontale: disinibizione, impulsività, euforia,ecc.
Corteccia temporale anteriore: ipersessualità, iperoralità, oltre a disturbi del linguaggio.

Diagnosi
Si ricorre alle stesse indagini dell'Alzheimer
Alla neuroimmaging ci sono della aree di atrofia evidente e dei pattern di compromissione della sostanza bianca che ad un occhio inesperto possono far pensare a un disturbo vascolare associato a malattia di Pick.
In questo caso le indagini funzionali sono ancora più importanti che nell'A. perché ci mostrano la distribuzione focale della neurodegenerazione.
Anche l'anatomia patologica ci evidenzia la forte discrepanza che c'è tra le circonvoluzioni frontali, fortemente atrofiche, e il resto degli emisferi.
Tra i pz con demenza fronto temporale che interessa spt il lobo temporale, e che quindi hanno prevalentemente disturbi del linguaggio, distinguiamo quelli con Afasia Primaria Progressiva:
Afasia Primaria Progressiva
In generale i disturbi del linguaggio si distinguono in 2 gruppi:afasia primaria progressiva e demenza semantica. Nel primo caso il soggetto ha un linguaggio disartrico con alterazione dell'organizzazione della parola di cui però ne capisce il significato: è anomico, disgrafico, dislessico e commette molti errori fonetici, non semantici. Tipicamente questa alterazione del linguaggio rimane isolata per almeno 2 anni o più, cosicchè il soggetto rimane solo afasico senza essere demente, tanto da continuare normalmente la propria vita e il proprio lavoro.
Difficoltà a trovare le parole (anomia)
Pattern di linguaggio anomalo
Deterioramento dello spelling
La diagnosi clinica è fatta:
quando le altre facoltà mentali (memoria per il quotidiano, abilità visuo-spaziale, comportamento), non appaiono grossolanemente compromesse
quando il linguaggio è la sola area di saliente e progressiva disfunzione per almeno i primi 2 anni
quando in neuroimaging non mostra altre specifiche lesioni che atrofia in regioni congrue con il disturbo del linguaggio
esame neuropsicologico indicativo (spesso può "esagerare" il reale deficit)
Nella demenza semantica il problema non sta nell'espressione ma nella comprensione del linguaggio:questi soggetti parlano, anche in modo fluente, ma non capiscono cosa stanno dicendo né quello che viene detto loro.
L'afasia può esordire con una confusione nel nominare oggetti appartenenti alla stessa categoria, es: il pz distingue tra una bottiglia e un finocchio ma non tra un finocchio e una carota. Questo perché la differenza tra grandi categorie linguistiche è ancora conservata, ma non quella tra le sottocategorie.
Degenerazione cortico-basale
Rientra tra i Parkinson plus, quindi si associano disturbi del movimento, e fa parte delle demenze fronto-temporali. La sua caratteristica è che si associano il danno corticale parietale(con i relativi disturbi) e quello sottocorticale extrapiramidale, cioè dei nuclei della base.
Sindrome acinetico-rigida asimmetrica progressiva ed aprassia ideomotoria
Degenerazione cortico-dentato-nigrica con acromasia neuronale
Segni corticali fenomeno dell'arto alieno
deficit della sensibilità corticale
mioclono
Segni da coinvolgimento dei nuclei della base bradicinesia
distonia
tremore
Decorso: progressivo
I sintomi tipicamente iniziano ad un arto superiore con rigidità, goffagine, incoodinazione. Spesso assume posture distoniche.
Successivamente aprassia d'arto
I deficit rimangono in genere confinati ad un arto per circa 2 anni, poi lento progressivo coinvolgimento degli altri, fino ad una severa generalizzata disabilità molti anni più tardi.
Una demenza clinicamente evidente non rientra tra gli aspetti precoci salienti, ma nel decorso della malattia prima o poi viene fuori un deterioramento fronto-temporale
N.B.: i diversi fenotipi clinici delle demenze sono riferibili agli stadi iniziali della malattia perché tutti i pz sono destinati ad una demenza globale!
Diagnosi
Studio elettroneurofisiopatologico (EMNG, PES, EEG, St. Magnet.): mioclono corticale
RMN/TC: atrofia corticale fronto-temporale asimmetrica e congrua con la clinica, atrofia dei nuclei della base, dei ventricoli laterali e peduncoli cerebrali, asimmetrica e congrua con la clinica
SPECT/PET: ipoperfusione/ipometabolismo in corteccia fronto-parietale gangli della base
"Non esistono markers biologici in vivo che permettano di porre diagnosi di certezza"
Nell'ambito delle demenze
fronto-temporali rientra
Paralisi Sopranucleare progressiva
Ĕ anch'essa una demenza con partecipazione del movimento, infatti rientra tra i Parkinson Plus e dà un coinvolgimento coticale(frontale) e sottocorticale(mesencefalico).
Criteri diagnostici:
È una forma neurodegenerativa caratterizzata da marcata limitazione della motilità oculare estrinseca(paralisi dei movimenti di verticalità) + Sindrome extrapiramidale prevalentemente assiale(per coinvolgimento dei nuclei della base) + Demenza frontale
Resistenza alla Levodopa
La neurodegenerazione interessa la substantia nigra, nucleo sub-talamico, globo pallido, tegmento ponto-mesencefalici, corteccia frontale, ippocampo
La degenerazione neurofibrillare è diversa dall'Alzheimer in quanto le fibrille sono rettilinee
Le
demenze fronto-temporali, ma più in generale tutte le Taupatie, non rispondono
bene agli anticolinesterasici, tranne
Alla terapia con anticolinesterasici si aggiungono i farmaci sintomatici per ridurre i disturbi del comportamento, i disturbi motori(anche se spesso sono resistenti all'L-dopa!), ecc. ed eventualmente farmaci sintomatici sperimentali
Demenza a corpi di Lewy
Si tratta di un'α-sinucleinopatia, infatti è dovuta all'accumulo dell'a-sinucleina, una proteina coinvolta nella degradazione delle proteine danneggiate.
Nella sua forma pura è rarissima infatti la demenza(che nella forma tipica è estremamente fluttuante nel corso della giornata) si accompagna sin dalle fasi precoci a segni parkinsoniani (bradicinesia, rigidità, mentre il tremore a riposo è raramente presente), anch'essi fluttuanti nell'arco della giornata.
1)Fluttuazioni (in particolare vigilanza e attenzione ma anche dei segni parkinsoniani)
2)La progressione di malattia è più veloce della m. di Alzheimer
3)Frequente presenza, anche in fase iniziale di malattia, di allucinazioni in particolare visive, ben dettagliate e reiterate(anch'esse fluttuanti)
4)I segni cognitivi di norma precedono i segni motori (bradifrenia, rallentamento psicomotorio), ma questo non sempre è vero.
5)Grande risposta alla terapia anticolinesterasica (importante)
6)particolare sensibilità ai neurolettici.
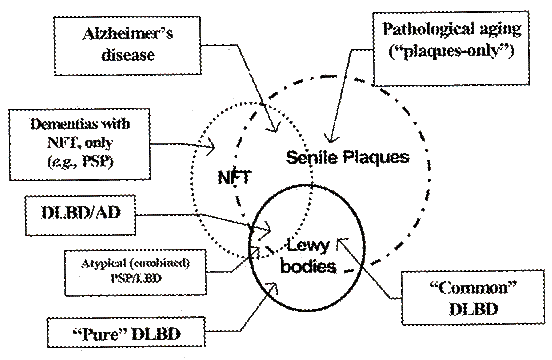
L'aspetto neuropatologico può essere importante per la diagnosi circostanziale di queste demenze ma allo stesso tempo può risultare poco definibile perché in uno stesso cervello ci sono molte sovrapposizioni tra i reperti: placche senili, corpi di Lewy, degenerazione neurofibrillare, ecc.
Questo diagramma ci serve per orientarci nella diagnosi differenziale e nella terapia.
|
|
| Appunti Nutrizione |  |
| Tesine Bellezza |  |
| Lezioni Bambini |  |