|
| Appunti superiori |
|
|
| Appunti superiori |
|
| Visite: 1850 | Gradito: |
Leggi anche appunti:La fogliaLA FOGLIA Le foglie con i rami formano la chioma di una pianta. Nella La gravidanzaErroneamente oggigiorno nel linguaggio comune , quando si parla di aborto, si intende Le vitamineLE VITAMINE Le vitamine sono composti organici essenziali, incluse tra |
 |
 |
BIOTECNOLOGIE DEI MICROORGANISMI
Biotecnologia: qualsiasi processo/intervento che comporti l'uso di un vivente o di parti di un vivente o prodotti metabolici di viventi (acino dell'uva, lievito.).
Le biotecnologie si distinguono in:
A- convenzionali: burro, impasti, vino ecc
B- genetico-molecolari: si basano sulla genetica e agiscono a livello delle molecole informazionali (DNA). Riguardano tutti i viventi. A noi interessano quelle microbiche, usate per riconoscere un MO a scopo analitico e scientifico e per creare nuovi individui. Es: la PCR serve per individuare MO isolati dall'ambiente, caratterizzarli e determinarne la presenza.
La caratterizzazione microbica si basa su due aspetti:
fenotipo (espressione del genotipo), che comprende la definizione di: caratteristiche morfologiche, metabolismo energetico, rapporti con l'ossigeno, esigenze nutrizionali, esigenze colturali, habitat, rapporti con gli altri esseri viventi, interesse dell'uomo, caratteristiche chimico-strutturali, immunologiche e sierologiche; come è formata la parete, le strutture interne ecc
genotipo: caratteristiche genetiche, individuate attraverso la genetica formale (Mendel) e la genetica molecolare.
Il genoma di una cellula serve a produrre le proteine, che sono l'espressione del fenotipo a livello molecolare. Le biotecnologie molecolari ci dicono quale ceppo è migliore per ottenere un determinato prodotto: ci dicono come modificare il fenotipo!
Tutti gli esseri viventi sono costituiti da:
polisaccaridi: polimeri di zuccheri semplici, primo fra tutti il glucosio. (amido, cellulosa.)
lipidi: sostanzialmente trigliceridi, costituiti da glicerolo e acidi grassi
proteine: polimeri di amminoacidi
acidi nucleici: DNA e RNA
Le proteine, oltre a C, H e O, contengono anche N e S.
La caratteristica importante comune a tutti queste molecole è di essere catene carboniose più o meno lunghe e ramificate. Il carbonio è l'unico elemento che si lega con se stesso. Vi si può paragonare solo il silicio, che però ha bisogno dell' O2 a fare da ponte.
Da un punto di vista elementare, quindi, l'uomo è uguale ai batteri: siamo fatti degli stessi atomi!
IL DNA
Il DNA è composto da monomeri, detti nucleotidi , uniti da ponti fosfato.
I nucleotidi sono costituiti da:
uno zucchero (ribosio nell'RNA o deossiribosio NEL dna, monosaccaridi: pentosi, aldosi).
una base azotata, ovvero una molecola ciclica, cioè con catena chiusa su se stessa a formare uno o due anelli. Adenina e Guanina sono purine, con 2 anelli; Citosina, Timina (o Gracile nell'RNA) sono pirimidine a 1 anello. Le basi sono legate allo zucchero covalentemente con un legame glicosidico.
una molecola di acido fosforico (H3PO4) che permette il legame tra gli zuccheri.
La base più lo zucchero costituisce il nucleoside.
Da notare la complementarietà delle basi azotate che si legano due a due, ma sempre una purina con una pirimidina: A-T con 2 legami H, C-G con 3 legami H.
Le basi azotate si trovano sulla parte interna del DNA, mentre sulla parte esterna si ha la sequenza ripetuta di zucchero-acido fosforico. Il legame tra zucchero ed acido fosforico si forma attraverso reazione tra 2 gruppi -OH delle 2 molecole: si perde un H2O e si forma il ponte ossigeno. L'acido fosforico usa due dei sui gruppo -OH per stabilire legami fosfodiestere, uno con il gruppo -OH attaccato al carbonio 3 di uno zucchero, l'altro con il gruppo -OH attaccato al carbonio 5 del secondo zucchero. Pertanto gli unici elementi variabili sono le basi azotate, mentre zucchero ed acido fosforico sono solo elementi strutturali che servono per tenere insieme il polimero.
Inoltre una estremità della catena porta un gruppo fosforico legato solo al carbonio 5 di uno zucchero, mentre all'estremità opposta troviamo uno zucchero con un carbonio 3 al quale non si attacca più nessun acido fosforico. Quindi un acido fosforico ha un'estremità 5' e una 3'. Nel costituire la doppia elica i due filamenti sono tenuti assieme da legami H.
GENOMA BATTERICO
I MO utilizzati nella genetica molecolare sono batteri procarioti perché sono organismi semplici che hanno solo riproduzione asessuata In questo modo si mantengono nel tempo le caratteristiche desiderate.
Caratteristiche genoma dei procarioti:
diametro del DNA di 1 mm=1/1000mm (1 cromosoma con più molecole di DNA, caricato negativamente). L'evoluzione microbica è stata limitata dalle piccole dimensioni della cellula, dentro cui non può essere contenuto il DNA di un eucariota. (1mm=10-3)
assenza di un nucleo organizzato,
unico cromosoma, che svolto è lungo 1 mm. Forma circolare. Avvolto attorno a molecole proteiche cariche positivamente. Assenti però gli istoni, caratteristici dei cromosomi eucarioti.
solo rare sequenze ripetute di DNA, che invece sono tipiche degli eucarioti.
non esiste la separazione in introni, parti che non vengono tradotte, ed esoni, parti che vengono tradotte. Invece negli eucarioti sono presenti introni ed esoni; durante la sintesi proteica, con la trascrizione a mRNA si mantiene la presenza di introni ed esoni, poi attraverso lo splicing si eliminano gli introni (mRNA corretto). Segue la traduzione a polipeptide.
presenza di trasposoni, frammenti di DNA che si spostano da un punto di un cromosoma, sito donatore, a un altro punto dello stesso (o magari di un altro se l'organismo è eucariote e ha più cromosomi), il sito bersaglio. Tale spostamento è chiamato trasposizione e determina dei cambiamenti nel cromosoma della cellula che possono anche portare alla morte della stessa o impedire l'espressione di un gene. In alcuni casi, invece, i portano della informazioni che sono utili. Nei batteri esistono due classi di trasposoni: quelli semplici, le sequenze di inserzione, che contengono solo sequenze necessarie alla loro trasposizione e i geni per le proteine che dirigono il processo, e i trasposoni complessi, che contengono anche dei geni soprannumerari rispetto a quelli necessari per la trasposizione.
nei MO sono possono essere presenti i plasmidi, tratti circolari di DNA extracromosomale a doppia elica e superavvolto. Sono autoreplicanti (hanno un pezzo di DNA che consente loro di duplicarsi indipendentemente dalla duplicazione del DNA cromosomale) e variano per le dimensioni. Portano diverse informazioni genetiche e sono responsabili di caratteri accessori. I plasmidi possono caratterizzare un ceppo, ma non una specie in quanto possono essere persi. Il plasmide più importante è quello sessuale, il plasmide F (della fertilità). I MO che lo possiedono sono F+ e hanno il pilo sessuale. Attraverso il pilo possono trasmettere il plasmidi agli F- che così diventano F+.
Le sequenze di inserzione e i plasmidi sono elementi trasponibili, pezzi di DNA che si muovono da un sito cromosomico ad un altro.
La molecola del DNA è stata studiata facendone l'analisi chimica. Importantissima è stata la scoperta delle diverse basi perché mi può dire se un determinato MO appartiene ad una data specie o no. Si è cercato di bypassare l'analisi chimica passando ad un approccio più biochimico.
Si esprime il contenuto % della coppia guanina-citosina per stabilire quali sono i rapporti in cui sono presenti le varie basi.
Il genoma batterico è stato sequenziato, ma ci sono ancora delle parti che non dicono nulla, che non si sa a cosa servano di preciso.
I geni reali dei MO e dell'uomo sono inferiori a quelli riportati perché è presente del DNA che non si esprime o non si sa cosa codifichi (soprattutto negli eucarioti). Questo non è DNA "spazzatura" ma è legato a modificazione di nostre malattie.
Alcuni MO sono patogeni obbligati, possono sopravvivere solo nell'ospite perché non sono in grado di sintetizzare alcune sostanze a loro necessarie. Questi MO hanno il genoma più corto contenente, quindi, meno informazioni. Ad esempio Rickettzia proweazekii è un patogeno obbligato dei pappagalli ed ha il genoma di 1 109 Da con 106 paia di basi (1milione 111mila 523: lo sappiamo con esattezza perché è stato sequenziato). I parassiti obbligati, che cioè possono vivere solo all'interno di un ospite, hanno bisogno di meno DNA.
Il cromosoma di E. coli presenta:
60% di DNA,
30% di RNA di trascrizione (mRNA)
10% di proteine. Queste per il 90% sono RNA polimerasi e per il 10% sono proteine caricate positivamente (spermina e spermidina) con ioni Ca++ e Mg++. Compensano le cariche negative del DNA e sostituiscono gli istoni che ci sono negli eucarioti.
Non ci sono istoni.
Il genoma batterico è costituito da 4 x 106 paia di basi=4000000
Sono presenti 600 molecole di mRNA, 100 di tRNA e 700 di rRNA.
NB: non si parla quasi mai di Dalton, ma di paia di basi, ovvero di nucleotidi.
Ogni gene codifica per una proteina.
Le proteine sono polimeri di aa. Ogni aa è codificato da una tripletta di nucleotidi.
Una proteina ipotetica ha 400 aa (PM=48000 Da).
400 * 3 (triplette di nucleotidi) = 1200 paia di basi. È un genoma medio.
Paia di basi del genoma totale / paia di basi di un gene = 4 106 / 1200 = 3300 geni
1 kbase di DNA a doppio filamento (ds) = 6.6 105 Da
1 kbase di RNA a singolo filamento (ss) = 3.4 105 Da
1 kbase di DNA codifica per una proteina di 333 aa.
Infatti un aa è definito di 3 nucleotidi, 1kb=1000 paia di basi e 1000/3=333.
Ovvero una proteina di 30000 Da ha bisogno di un DNA con 810 paia di basi.
![]()
![]()
![]() organismo PM DNA (Da) kbasi = 1000 paia di basi di DNA
organismo PM DNA (Da) kbasi = 1000 paia di basi di DNA
E. coli 2.6 109 4 103 (senza plasmidi)
S. cerevisiae 1.3 1010 2 104 (ha più cromosomi)
Uomo 3.6 1012 5.6 106 (46 cromosomi)
Le dimensioni degli enzimi dipendono dal tipo di cellula, di MO e dalla funzione della proteina, ma non è detto che la dimensione della proteina sia in funzione della dimensione della cellula:
- la b-galattosidasi di E. coli = 1173 aa
- l'insulina di bue = 51 aa
Le proteine si differenziano per tipo di aa, lunghezza, disposizione di aa. Sono serin e tiol proteasi, con diversi inibitori.
La b-galattosidasi scinde il lattosio per ricavarne energia.
Anche l'uomo posside questo enzima ma diverso, sia per composizione in aa, che per sequenza e rapporto fra i diversi aa.
L'insulina di bue è una molecola piccola. È stata il primo prodotto della biosintesi molecolare e non è stato dunque più necessario uccidere numerosi animali. È stato un processo facile proprio perché la molecola è piccola (150 nucleotidi circa).
Quindi per descrivere un organismo o un microrganismo dobbiamo partire dal suo fenotipo, che non è altro che l'espressione del suo DNA.
MODIFICHE DEL GENOMA
Prima degli anni '50, per ottenere animali con caratteristiche desiderate, si modificava solo il fenotipo, agendo sull'individuo, oppure si modificava il genotipo attraverso incroci tra animali della stessa specie o di specie diverse.
Nel 1944 Avery, Mc Donald e Mc Carty (Watson e Creek) determinarono la struttura del DNA.
Dopo gli anni '50 si sono definiti i MO studiando il genotipo; si può modificare il genoma microbico attraverso una mutazione o una ricombinazione genetica, ottenendo degli OGM.
Clonazione = creazione di un nuovo vivente senza riproduzione sessuata; il nuovo ha le stesse caratteristiche dell'individuo di partenza. Le talee sono cloni vegetali.
RICOMBINAZIONE GENETICA
È la capacità dei batteri di acquisire DNA estraneo (nudo) e può avvenire anche naturalmente. Con questo metodo si effettua una modificazione mirata e, da un'analisi di indagine, si passa a creare sonde nucleiche/nucleotidiche contenenti un tratto di DNA caratteristico di un determinato MO. Le sonde sono utilizzate per individuare un MO in una matrice anche complessa (alimento), per determinare se due MO sono uguali o diversi e identificare nuovi MO (non coltivabili). Sono usate per la caratterizzazione dei MO; infatti, se trovo che un MO ha una caratteristica particolare, posso sfruttare tale caratteristica per trovare il MO nell'alimento.
Oggi conosciamo il 10% dei MO presenti sulla terra. Con le sonde possiamo scoprire MO nuovi che non abbiamo mai visto o che non vedremo mai perché non riusciamo a coltivarli. (forse va messo da altra parte. nn centra con ricombinazione)
La ricombinazione può avvenire con tre modalità:
1- trasformazione: processo attraverso il quale una molecola di DNA viene trasferita ad una cellula competente. (Griffith, 1928).
Lo Streptococcus pneumonie è un MO patogeno presente in due forme (entrambe patogene): forma L e forma R. La forma L ha colonie lisce poiché le cellule sono ricoperte da una capsula mucosa ed è virulenta perché sfugge ai meccanismi di difesa del nostro corpo. La forma R è quella rugosa in quanto priva di capsula e non è virulenta perché rimane vittima dei nostri meccanismi di difesa.
Griffith ha inoculato in diversi topi:
la forma L e il topo è morto
la forma R e il topo è sopravvissuto
la forma L uccisa col calore e il topo è sopravvissuto
sullo stesso topo la forma R e il topo è morto
Da quest'ultimo topo ha estratto un ceppo capsulato, con caratteristiche sia della forma R che L, quindi ha affermato che il MO si è trasformato a causa di un "principio trasformante", il DNA. La capacità del MO di incorporare DNA estraneo è attiva solo se le cellule si trovano in un certo stato, detto di competenza; ad esempio, se nel mezzo è presente del calcio. Infatti, non tutte le cellule e non in qualunque situazione sono in grado di acquisire DNA.
Nel 1944 Avery, Mc Donald e Mc Carty hanno stabilito la natura del principio trasformante scoprendo il DNA.
2-coniugazione: processo nel quale il trasferimento di DNA avviene attraverso il contatto tra cellula donatrice e cellula ricevente. È un processo unidirezionale (Lederberg & Tatum, 1946).
Lederberg e Tatum hanno ripetuto l'esperimento di Griffith su E. coli. Hanno usato un tubo ad U con un setto permeabile al DNA ma non alle cellule, che separa i due bracci ed evita il contatto tra i due ceppi microbici. La trasformazione è stata confermata su S. pneumonte, ma non su E. coli.
Togliendo il setto dal tubo a U i due ceppi di E. coli entrano in contatto e producono un ceppo con caratteristiche di entrambi. Questo può avvenire perché un ceppo è F+ (cellula donatrice con fertilità positiva, cioè produce il pilo sessuale) e l'altro F- (fertilità negativa, cellula ricevente priva del pilo). Le due cellule entrano in contatto, la cellula F+ duplica il DNA plasmidiale che codifica per il pilo F e questo permette il passaggio unidirezionale del materiale genetico all'altra cellula.
Sono noti due tipi di cellule donatrici: F+ e ceppi HFR. Nel primo caso (F+) la cellula ricevente diventa F+ perché ha acquistato il plasmide del pilo (che rimane in forma CCC). Il plasmide esiste in forma autonoma ed è in grado di trasferire solo se stesso Nella coniugazione ad alta frequenza, HFR, il plasmide si integra nel DNA cromosomale; in questo modo al MO successivo potrà passare solo una parte del gene che codifica per il pilo F e un tratto di DNA cromosomale. Se ciò si verifica il MO può non acquistare la capacità di produrre il pilo (non tutte le cellule diventano F+) ma altre caratteristiche. Il plasmide integrato è in grado di mobilizzare il trasferimento di porzioni di DNA cromosomale.
Dopo che le cellule hanno preso contatto inizia la sintesi di DNA e un singolo filamento viene trasferito dal donatore (F+ o HFR) al ricevente F-. La sintesi del DNA sia nel donatore che nel ricevente assicura che venga mantenuta in entrambe le cellule la struttura a doppia elica e che nessuna informazione genetica del donatore venga perduta. Sebbene i ceppi HRF trasferiscano geni cromosomali ad alta frequenza, non sono in grado di convertire cellule F- in F+ o HRF, poiché il trasferimento dell'intero cromosoma avviene solo raramente dato che le cellule si staccano prima che il processo sia completato. Al contrario le cellule F+ convertono efficacemente F- ad F+ grazie alla natura trasmissibile del plasmide.
Attenzione: la coniugazione non è un processo di trasformazione perché la presenza di un setto permeabile alle macromolecole (DNA) ma non alle cellule dimostra che il contatto cellula-cellula è condizione necessaria per la ricombinazione.
3-trasduzione: trasferimento di DNA da un MO ad un altro per intervento di un fago temperato, ovvero con ciclo lisogeno (Zinder & Lederberg, 1952). Studi su Salmonella tiphimurium hanno dimostrato che l'acquisizione di DNA estraneo può essere veicolata da fagi che hanno il ciclo lisogenico.
La trasduzione può essere :
MUTAZIONE
Una mutazione consiste nella modificazione del DNA durante la sua ricostruzione a seguito di danni subiti, non è una modificazione diretta delle basi causata dall'esposizione all'agente mutageno. Le mutazioni avvengono raramente.
Le mutazioni possono essere naturali (spontanee) o indotte in laboratorio con agenti chimici (benzene, acido nitroso, nitroso guanidina, analoghi delle basi, bromuro di etidio) o agenti fisici (raggi UV, radiazioni ionizzanti); si genera una modificazione del genotipo che non è prevedibile (è casuale) e non è sempre favorevole (può portare anche a morte cellulare).
Quando una cellula subisce delle alterazioni al DNA, si attivano dei meccanismi per ripararlo; si attivano gli enzimi di restrizione (endodesossiribonucleasi) che tagliano il DNA a doppia elica in punti precisi, cioè dove sono presenti delle sequenze palindrome (speculari). Per evitare che gli enzimi taglino altro DNA, alcuni nucleotidi vengono metilati.
Gli enzimi di restrizione agiscono solo su DNA a doppio filamento e servono anche per eliminare DNA estraneo (di fagi). Il taglio asimmetrico dell'enzima di restrizione genera delle estremità appiccicose (sticky end) sfruttate dalle ligasi che creeranno un nuovo legame tra le estremità del DNA idrolizzato (può anche tagliare in modo simmetrico).
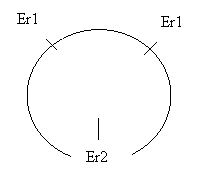
Il primo enzima di restrizione è stato estratto da E. coli ed è Eco R1 (E da Escherichia, co da coli, R dal ceppo e 1 perché è il primo Er estratto da questo ceppo) il quale riconosce la sequenza palindroma seguente e taglia il legame tra A e G.
![]()
![]()
![]()
![]()
![]() G-A-A-T-T-C G-G-A-T-C-C G-G-C-C
G-A-A-T-T-C G-G-A-T-C-C G-G-C-C
![]()
![]()
![]() C-T-T-A-A-G C-C-T-A-G-G C-C-G-G
C-T-T-A-A-G C-C-T-A-G-G C-C-G-G
Da Bacillus amiloliquefaciens è stato estratto il B am H1 e da B. licheniformis il B li I. Quest'ultimo non genera estremità asimmetriche e per utilizzarlo si devono attaccare alle estremità tronche delle sequenze note per creare l'asimmetria riconosciuta dalle ligasi.
ISOSCHIZOMERI: enzimi di restrizione uguali ma appartenenti a diversi MO, quindi tagliano diversi tratti di DNA in corrispondenza della stessa sequenza.
ESTRAZIONE E PURIFICAZIONE DEL DNA
Innanzitutto bisogna avere il ceppo su cui operare, che deve essere rivitalizzato, isolato e controllato.
Procedimento:
Coltivazione in un opportuno terreno
Centrifugazione per separare la biomassa, che rimane sul fondo
Lavaggio biomassa con acqua distillata.
Lisi cellulare: si sospende in tampone SSC (salincitrato) e si procede in diversi modi a seconda dei casi. Se il MO è Gram+ si utilizza il lisozima, che attacca i legami del mucopeptide della parete cellulare facendo scoppiare la cellula. Oppure si coltiva il MO in presenza di penicillina, attiva contro i Gram+. In questo caso però bisogna avere l'accortezza di coltivare il MO in ambiente isotonico, in modo che la pressione interna ed esterna si equivalgano (aggiungo zucchero), così si formano sferoplasti privi di parete che a contatto con SSC scoppiano. In alternativa posso usare una pressa (French press) in cui si crea un aumento di pressione con un sistema a pistoni che obbliga il batterio a passare in un foro di 0.2 mm di diametro, cioè decisamente più piccolo della cellula.
Aggiunta di EDTA (etilendiamminotetracetato) che chela gli ioni bivalenti (calcio) che stabilizzano le endodesossiribonucleasi (DNAasi, enzimi che tagliano il DNA), ottengo una massa gelatinosa, vischiosa e non liquida, perché predominano le macromolecole. Il Ca++ è indispensabile alle DNAasi per lavorare; chelandolo impedisco che queste rompano il DNA.
Aggiunta di SDS (o lauril solfato) che denatura le proteine liberando la molecola di DNA. La massa gelatinosa diventa più trasparente (purificazione). Ottengo una massa biancastra.
Per separare i diversi componenti della massa e quindi purificare il DNA, si aggiunge un ugual volume di cloroformio o etanolo (rapporto 1:1) e si agita.
Si centrifuga a 8000giri/min e si ottenengono 3 strati: il più basso è solvente (cloroformio) contenente lipidi e sostanze apolari (limpido giallino); il centrale contiene proteine denaturate, cellule intere e rotte (bianco); il surnatante è una soluzione acquosa contenente DNA, RNA, polisaccaridi e proteine solubili.
Il surnatante viene separato e posto in un cilindro con etanolo freddo al 95 % in quantità pari al doppio della soluzione acquosa (2 volumi). L'alcol si stratifica sopra il surnatante.
Si introduce una bacchetta pulita nello strato di etanolo e si mescola lentamente; l'alcol scende e rende insolubili DNA, RNA, polisaccaridi e proteine che si attaccano alla bacchetta,. rimangono in soluzione solo le parti rotte, troppo piccole per attaccarsi.
Ciò che si è separato sulla bacchetta viene fatto asciugare; il DNA diventa bianco e viene risospeso in un tampone salin-citrato (SSC=NaCl più citrato di Na). Si ripete l'estrazione più volte fino a quando la fase intermedia è assente, cioè si sono separate tutte le cellule rotte e le proteine. Rimangono solo DNA, RNA e polisaccaridi.
Si scioglie nuovamente il DNA e si aggiungono RNAasi che idrolizzano l'RNA e si ripetono le fasi di purificazione per eliminare RNAasi, che sono delle proteine. Operazione preventiva: riscaldamento a 80°C per 10' per eliminare eventuali DNAasi.
Si ultracentrifuga a 30000 giri la soluzione contenente solo DNA e polisaccaridi (bilanciata anche la densità); il DNA rimane in soluzione e sul fondo della provetta si trovano i polisaccaridi in una massa gelatinosa.
Sul DNA puro si legge il % GC allo spettrofotometro.
determinare la quantità di DNA
Si può effettuare una lettura allo spettrofotometro per determinare la concentrazione di DNA.
Il DNA ha un picco con massimo a 260 nm (le basi assorbono a questa lunghezza d'onda), ovvero nell'UV. Anche le proteine e l'RNA assorbono all'UV, ma le proteine hanno il massimo di assorbimento a 280 nm e l'RNA a 240 nm. Per fare l'analisi spettrofotometrica non si possono usare le normali cuvette di vetro, in quanto il vetro non lascia passare i raggi UV; è necessario utilizzare delle cuvette di quarzo, molto costose.
N.B. quando il DNA assorbe le radiazioni UV viene da esse danneggiato. Quindi mai fare le lampade abbronzanti!
L'analisi spettrofotometrica mi dà un'idea della quantità di DNA. Attenzione però: alcune proteine assorbono a 260 nm e DNA e RNA assorbono a lunghezze d'onda simili, quindi le curve in parte si sovrappongono.
N.B. il DNA a doppio filamento assorbe meno di quello a singolo filamento: infatti i legami H tra le basi sui filamenti opposti del DNA ds riducono l'abs delle radiazioni UV.
La dissociazione del DNA è ottenibile agendo su:
Temperatura (85-96 °C)
concentrazione del tampone
alta concentrazione di urea
pH > 11 e pH < 3
Ad esempio alzando la temperatura si ottiene la denaturazione del DNA che poi riassocia spontaneamente con il raffreddamento.
%GC
Nel 1964 gli studiosi Marmur e Doty hanno individuato un sistema biochimico per determinare il %GC sul totale delle basi, basato sul fatto che il DNA assorbe a 260 nm. Il %GC è una misura diretta che permette di ottenere indicazioni sul DNA del ceppo in analisi.
Il grafico riporta la densità ottica a 260 nm in funzione della temperatura e si registra leggendo l'assorbanza di un campione di DNA posto in una cuvetta di quarzo durante un graduale e lento riscaldamento. Ciò che si osserva al raggiungimento di una certa T (tra 85 e 95°C) è una ipercromicità, cioè un aumento di assorbimento, associato alla denaturazione del DNA che passa da un filamento a doppia elica (ds) a due filamenti a singola elica (ss). Raggiunto il massimo di assorbimento la curva riprende con un andamento piatto, parallelo all'asse x.
La temperatura di denaturazione (Tm=melting temperature) è il valore di temperatura a cui metà del DNA è denaturato; dipende dalla composizione in nucleotidi del DNA, ovvero dalla composizione % nelle diverse basi. In particolare è risultato che maggiore è il %GC, più è stabile la molecola di DNA e maggiore è quindi il suo punto di fusione. Infatti, nel DNA a doppia elica le coppie CG sono unite da 3 legami H, mentre le coppie AT solamente da 2. Quindi il DNA con un contenuto maggiore di CG avrà più legami H e i suoi filamenti si separeranno a temperature più elevate. (N.B.: i lavori erano condotti su E. coli)
Il %GC si ricava dal Tm con la formula:
Tm - 69.3
![]() %GC =
%GC =
0.41
dove:
69.3 è l'intervallo di temperatura necessario per la denaturazione del DNA; è caratteristico di ogni MO (E. coli) e dipende dal tampone usato.
0.41 è il coefficiente angolare della retta in salita.
Il metodo del %GC è usato per differenziare due specie: se due MO hanno un %GC che differisce di 3-4 unità potrebbero essere della stessa specie, se differiscono di più di 7-8 unità sono di due specie diverse. Gli esperimenti sono stati condotti su E. coli, che ha %GC = 50% (varia a seconda del ceppo); il valore è stato ottenuto dal confronto con l'analisi chimica. Se un altro MO ha %GC = 50 potrebbe appartenere a E. coli, ma se è diverso da 50% (per più di tot punti) si è sicuri che non è un E. coli.
Facciamo qualche esempio di lettura delle curve.
Esempio 1.
caso A: retta con angolo minore di 0,41.
La curva ci dice che siamo in presenza di DNA sporco di qualcosa che assorbe a 260 nm, cioè proteine e RNA. Infatti questi possono assorbire a 260 nm modificando la pendenza della curva.
caso B: curva piatta.
In questo caso abbiamo a che fare con DNA già denaturato, oppure proteine o ancora RNa singolo.
Esempio 2.
Stessa inclinazione ma diverso Tm.
Questa curva invece ci dice che siamo in presenza di due DNA uguali immersi in due tamponi con differente forza ionica. È differente la concentrazione della soluzione in cui è immersoli DNA. Maggiore è la forza ionica (maggiore concentrazione del tampone), maggiore è la stabilità della molecola, perché i sali in soluzione complessano il DNA e sono quindi necessarie maggiori temperature per denaturarlo. Se invece sono due DNA diversi il secondo ha un maggiore % CG per cui la curva è spostata a destra. N.B. i tamponi sono fatti di acidi deboli e basi deboli, quindi in questo caso ho sbagliato la concentrazione di basi.
Esempio 3.
Lavoro con lieviti (eucarioti).
Il DNA degli eucarioti si può presentare con questo andamento perché oltre al DNA nucleico, vi è anche quello mitocondriale. Quest'ultimo si denatura a temperatura più bassa perché ha un %GC inferiore; protraendo il riscaldamento si ha la denaturazione anche del DNA del nucleo. Quindi la prima parte della curva corrisponde a DNA mitocondriale, sempre più basso, mentre nella parte più alta troviamo il DNA nucleico. È proprio questa parte della curva che dobbiamo considerare per determinare Tm e quindi %GC.
Gli attinomiceti hanno un alto %GC, maggiore di 60%, quindi il Tm è spostato verso destra. Di conseguenza il campione in analisi ha bisogno di temperature maggiori di 90°C per denaturarsi. Può arrivare anche a 100°C, temperatura di ebollizione dell'acqua. Dato che il tampone è acquoso una eventuale evaporazione dell'acqua crea non pochi problemi (si concentra). È necessario abbassare Tm diminuendo la forza ionica del tampone (aumentando la forza ionica ho uno spostamento della curva verso destra): anziché usare un tampone 10 molare se ne usa uno 1 molare (tra l'altro fosfato).
Durante la lettura del %GC è importante controllare:
il tampone (influenza il Tm)
la conducibilità
presenza di errori umani e di strumentali
perché basta che lo strumento legga una temperatura superiore di 1°C per far variare il %GC.
Queste fonti di errore si eliminano utilizzando uno standard interno, che è un DNA noto (cioè di cui già si conosce il %GC) di E. coli: si confronta il risultato dello standard con quello del campione.
In questo modo la formula diventa:
% GCx = % GCE.coli + 2.44 (Tmx - TmE.coli)
2.44: 1/0.41; varia in funzione della concentrazione
%GCE.coli: venduto liofilizzato (50%)
Tmx: Tm del campione
TmE.coli: Tm di E.coli, noto
Il controllo interno permette, nel caso per esempio sia avvenuto un errore nella preparazione del tampone, di ripetere l'errore nei due casi, e quindi di annullarlo. Idem nel caso in cui lo spettrometro sia "sballato". Non posso mai sbagliare!
Il confronto si basa esclusivamente sul Tm, in questo modo si valuta solo il Tm e non la purezza del DNA.
Come abbiamo già detto, il %GC ci può servire per classificare i MO confrontando i %GC di diversi MO con quello del ceppo di riferimento. Ci fornisce anche informazioni sulla composizione proteica.
Attenzione: ci possono essere MO con lo stesso %GC che sono però MO diversi. Possiamo avere solo la certezza che due MO appartengono a specie diverse nel caso in cui c'è una differenza in %GC di + o - 4 punti percentuali.
Ci occorrono altre informazioni, ad esempio sulla sequenza di basi, per fare confronti migliori tra MO specie e ceppi.
OMOLOGIA GENETICA
Che cosa succede raffreddando il DNA? Le due molecole si riassociano, ma solo quelle omologhe, cioè complementari l'una all'altra. Il risultato è l'ibridazione di due molecole uguali o tuttal più leggermente diverse.
Si parla di (4 nomi per lo stesso fenomeno; evoluzione nel tempo):
riassociazione molecolare DNA-DNA
ibridazione molecolare DNA-DNA
grado o % di omologia genetica
grado di similarità delle sequenze polinucleotidiche, il più corretto perché non si può studiare tutto il DNA, che è troppo lungo, ma solo alcune sequenze.
Si verifica raffreddando un campione di DNA precedentemente riscaldato per separare i filamenti; i filamenti si associano solo se sono complementari. Si effettua utilizzando un ceppo type di riferimento e un ceppo incognito e si valuta se i due filamenti ibridano; se ibridano sono dello stesso ceppo o hanno alta omologia, altrimenti appartengono a due MO diversi.
I due metodi (radioattivo e spettrofotometrico) sono uguali, quindi in entrambi si deve tener presente il tempo e la concentrazione di DNA.
1° sistema competitivo/metodo radioattivo:
Per determinare se un MO appartiene ad una certa specie ci si basa su studi di competizione tra il DNA del ceppo incognito (x) e DNA ceppo type, di riferimento.
Di entrambi i ceppi si fanna delle colture a freddo e si usano:
-DNAtype: denaturato (ovvero ss)
rotto e denaturato
rotto, denaturato e marcato
-DNAx: rotto e denaturato
Per ottenere il DNA marcato aggiungo alla coltura adenina o gracile radioattivi
Nb: è il C dell'adenina o dell'uracile che è radioattivo.
Procedimento:
Si preparano le due colture a freddo: una per il ceppo type e l'altra per il ceppo type marcato, si estrae il DNA e lo si purifica. Si ottiene DNA type a doppio filamento.
Si denatura il DNA con NaOH o KOH diluito ottenendo i singoli filamenti.
Il DNAt ss denaturato viene fissato su un supporto di nitrocellulosa attraverso una filtrazione a freddo (N.B. tutte le molecole organiche si lavorano a freddo) overnight (18h). La soluzione da filtrare è diluita in modo che il DNA si disponga sulla membrana ben separato. La filtrazione ha proprio lo scopo di tenere separati i DNA.
La membrana di nitrocellulosa viene trattata con polivinil pirrolidone che occupa il filtro dove non è presente DNA.
Il filtro viene essiccato (con lampade IR) e con un foratappi si creano tanti dischetti che contengono DNA type denaturato (intero e non marcato).
Si prendono 3 vials (provette a fondo piatto) e in ognuna si aggiunge un dischetto di DNA type a singolo filamento, a lunga catena e la soluzione di DNA type rotto, denaturato e marcato. Nel 1° non aggiungo nient'altro. Nella 2° vial si aggiunge anche il DNA type rotto, denaturato e non marcato. Infine nella 3° si aggiunge il DNA rotto e denaturato del ceppo incognito.
Addiziono il tampone e incubo overnight a bagnomaria a T=Tm-25°C, in leggera agitazione. La bassa temperatura permette la riassociazione o ibridazione degli omologhi.
Si lava il dischetto da tutto ciò che non si è legato e per effettuare la lettura si aggiunge un liquido di scintillatore e con vials speciali si pone in una apparecchiatura, il contatore Gaigher, che rileva la radioattività espressa in colpi/min. Attraverso questo valore, grazie ad una formula, si calcola il grado di omologia genetica.
Nella 1° vial si ha MAX radioattività perché non ci sono competitori.
Nella 2° la radioattività è MIN perché c'è la massima competizione tra DNA type e se stesso (marcato e non marcato).
Nella 3° vial (sistema eterologo) c'è competizione tra DNA type e ceppo incognito e la radioattività dipende dall'omologia tra i due DNA: più sono omologhi e minore è la radioattività. Se il ceppo x è lo stesso è minima.
La biodiversità a livello di specie fa si che ci siano gradi di omologia variabili; si considerano appartenenti alla stessa specie quei ceppi che hanno tra loro omologia genetica di 70-100 %. Quindi ceppi della stessa specie possono presentare una omologia anche inferiore al 100%! È la dimostrazione numerica della biodiversità genetica nella specie.
Si tratta dunque di un sistema valido per determinare se due specie sono uguali.
Lo svantaggio consiste nell'utilizzo dei marcatori radioattivi che sono costosi, soprattutto nello smaltimento, e pericolosi. Inoltre tale sistema, per quanto valido, risulta un po' laborioso.
cpmSOsc (1°) - cpmSEcc (3°)
% DI OMOLOGIA GENETICA = 100
cpmSOsc (1°) - cpmSOcc (2°)
cpm = colpi per minuto
SO = sistema omologo: ceppo di riferimento, type
SE = sistema eterologo: ceppo type più ceppo x (3)
sc = senza competitore (max radioattività) (1)
cc = con competitore (min radioattività) (2)
Se cpmSEcc è alto si ha poca affinità.
Esempio:
4000 - 3000
![]() * 100 = 33
% è l'indice di biodiversità dei batteri
* 100 = 33
% è l'indice di biodiversità dei batteri
4000 - 1000
2° sistema competitivo/metodo spettrofotometrico:
Per valutare l'omologia genetica si sfrutta il fatto che due filamenti omologhi abbassando la temperatura del campione riassociano e dunque si modifica anche l'assorbimento all'UV.
La competizione si ha tra ceppo type, con una certa curva di denaturazione, e ceppo ignoto, di cui si vuole determinare la specie, con la propria curva di denaturazione. La curva relativa alla miscela dei due può non esistere se le due molecole non hanno affinità.
Nota bene:
la denaturazione è favorita da T>Tm e da una bassa concentrazione salina (riduco il tampone)
la riassociazione invece è favorita da T<Tm e da un'alta concentrazione salina
Inoltre il grado di riassociazione può essere influenzato dalla quantità di DNA e dal tempo. Infatti, perché il sistema sia competitivo, la C di DNA deve essere uguale (se no la seconda curva varia) e anche i tempi devono essere precisi (50% riassociazione). Dunque capiamo che le condizioni devono essere stringenti: obbligo a riassociarsi solo i filamenti veramente omologhi. È ad esempio una condizione stringente una alta C salina e T=Tm.
Condizioni operative:
tampone con alta molarità (5 M), per favorire la riassociazione. Stringenza alta
T di riassociazione Tr=Tm-25°C (per passare da ds a ss)
spettrofotometro con sistema di riscaldamento progressivo, 3 cuvette (di quarzo, per leggere l'UV) e lettura simultanea
software per i calcoli
Procedimento:
1) Preparazione del saggio e lettura allo spettrofotometro. Si legge la DO e si ricava la quantità di campione:
1° cuvetta: DNA puro del ceppo type, 75 mg/mL (DO = 1.5)
2° cuvetta: 75 mg/mL di ceppo x (DO = 1.5)
3° cuvetta: miscela di DNA type e DNA del ceppo x in rapporto 1: 1 (ciascuno in 37.5 mg/mL), la somma dei DNA deve essere 75 mg/mL, così c'è competizione.
DO = 1 se C=50 mg/mL 1 mg/mL=
2) Denaturazione a 98°C per 5-7 minuti, in funzione del ceppo.
3) Riassociazione alla Tr per 30-40 minuti.
La DO iniziale è pari a 1.5 (75 mg/mL); scaldando il DNA il filamento si dissocia e la DO incrementa fino a rimanere costante (DO max). Abbassando la temperatura anche la DO si abbassa perché i filamenti riassociano; a T=Tr=Tm la DO è pari a DO50.
Per le tre provette (type, ceppo x, miscela) si calcolano:
DO = DO max - DO iniziale
DO50 = DO iniziale + ½ DO
½ Cot = ½ DO50 * t
t = tempo in ore per avere il 50% di riassociazione
½ Cotmix + ½ Cot100 - ½ Cot0
% RIASSOCIAZIONE = 1 - * 100
½ Cot100
½ Cotmix = ½ DO (al 50% riassociazione) della terza provetta * tempo (h)
½ Cot100 = media dei ½ Cot dei DNA omologhi (sia ceppo type che x, dipende dall'affinità)
½ Cot0 = somma dei ½ Cot dei DNA omologhi (dipende dall'affinità tra loro)
Se % di riassociazione è superiore al 70% i due MO appartengono alla stessa specie.
I fattori che influenzano il processo sono la temperatura, il tempo e la concentrazione.
La denaturazione è favorita da T>Tm e bassa concentrazione salina, mentre la riassociazione è favorita da T<Tm e da un'alta concentrazione salina. Il processo è anche influenzato da: quantità di DNA e tempo; quindi è necessario lavorare con concentrazioni di DNA precise (uguali per i due filamenti) e seguendo certi tempi.
Al variare della temperatura, i primi due campioni faranno registrare una certa curva di denaturazione e riassociazione (uguali o diverse tra loro); il terzo campione può riassociare subito, se i due DNA sono omologhi, oppure dopo un certo tempo perché ogni filamento di DNA deve ritrovare il proprio omologo.
Se ceppo type e ceppo x sono uguali o hanno alta omologia, la variazione di DO ha lo stesso andamento e i filamenti si scambiano tra loro. Hanno più o meno la stessa curva.
Nel secondo grafico i due filamenti di DNA non sono omologhi ed è necessario del tempo perché ogni filamento trovi se stesso (il proprio complementare) per riassociare; i due ceppi sono diversi ed hanno diverso Tm, Tr e %GC.
Questo metodo è utilizzato dopo aver calcolato il %CG dei due ceppi poiché non è detto che due ceppi con %GC uguale o che differisce di 3-4 unità siano omologhi; con questo metodo si ottiene un valore altamente discriminante. Se invece due ceppi hanno valore di %GC differente significa che sono diversi e non è necessario usare il metodo spettrofotometrico (un ceppo con 55% e l'altro con 62%).
È un metodo nato negli anni sessanta, ma ancora valido. Permette di ottenere gli stessi risultati già dati dai metodi tradizionali. Cioè si ha la dimostrazione della stretta correlazione tra genotipo e fenotipo. Ad esempio pensiamo al fatto che per differenziare le specie si sfruttano soprattutto le esigenze nutrizionali. Ma queste dipendono dagli enzimi, che sono fatti di proteine. Da determinativo a sistematico (c'è anche filogenesi).
NB: se conosco il %GC, quando decido di calcolare l'omologia genetica e quando no? Se ho %GC tra i due ceppi e so che non sono la stessa specie, allora è inutile fare tutto questo.
ENZIMI DI RESTRIZIONE
I MO hanno dei meccanismi di riparazione del DNA, ad esempio in seguito a trattamento con UV, che hanno come base gli enzimi di restrizione. Questi enzimi hanno la capacità di tagliare il DNA in sequenze precise, endodesossiribonucleasi (DNAasi). Ci sono poi altri enzimi in grado di legarlo, ligasi (taglio e cucito).
Quando un enzima di restrizione taglia il DNA cromosomale crea dei frammenti a diverso PM che non possono essere analizzati allo spettrofotometro; per la separazione si utilizza una gel-elettroforesi. Per le proteine si usa una matrice di acrilammide, mentre per separare il DNA è necessario utilizzare un gel di agarosio perché ha maglie più larghe, idonee al passaggio delle molecole di DNA (DNA: 1x106, proteina: 40000).
N.B. lavorano sempre su DNA ds.
Con questa tecnica i frammenti di DNA si separano in base al peso molecolare per applicazione di un campo elettrico (migrano dall'anodo al catodo in quanto il DNA ha carica negativa). I profili elettroforetici dei prodotti di digestione degli enzimi di restrizione sono analizzati per confronto con quelli ottenuti da DNA noti. La visualizzazione avviene per esposizione a luce UV in presenza di Bromo di etidio (cancerogeno) che evidenzia le bande con una colorazione arancione.
Esempio: Lactobacillus helveticus e jucuntis hanno lo stesso % GC, ma un diverso uso del lattosio, caratteristiche confermate dal profilo elettroforetico.
Gli enzimi di restrizione non sono usati sull'intero DNA perché si originerebbero molti frammenti non analizzabili; sono invece utilizzati facilmente sui plasmidi perché questi sono piccoli.
RFLP: Restriction Fragment Length Polymorphysm Analysis
Mette in evidenza il polimorfismo tra il DNA di diversi MO appartenenti ad una stessa specie.
Risultato: ottengo 2 frammenti di DNA. Se li metto vicini quelli complementari si riassociano. Se attacco una molecola di DNA con gli enzimi di restrizione che mi danno dei frammenti asimmetrici posso spontaneamente attaccare un pezzo di DNA con un altro: faccio un ibrido. Poi attacco il tutto con le ligasi.
Isoschizomero: uno stesso enzima di restrizione, però prodotto da due batteri diversi che riconosce sempre la stessa sequenza.
PLASMIDI
Il DNA plasmidiale è l'1-3 % del genoma batterico (il Bacillus ha un solo grosso plasmide).
I plasmidi sono autoreplicanti, cioè sono in grado di duplicarsi indipendentemente dal DNA cromosomale, e contengono diverse informazioni genetiche utili al MO, come la resistenza agli antibiotici o la produzione di enzimi, o in alcuni la patogenicità.
I plasmidi non hanno nessuna omologia con il DNA cromosomale.
Nei plasmidi possono essere presenti geni che codificano per:
enzimi del metabolismo del lattosio (acidificanti) es. L. lactis,
enzimi del metabolismo del galattosio (acidificanti)
produzione di proteasi (idrolisi della caseina),
resistenza agli antibiotici (sviluppo in presenza di antibiotici nel latte. Sono i plasmidi R, coniugativi perché possono trasferire la resistenza a ceppi sensibili attraverso il contatto tra le cellule),
enzimi del metabolismo dell'acido citrico (aromatizzanti),
batteriocine (equilibri biologici) es. nisina (lantibiotici) non resistenze nei MO (patogeni e non) perché viene digerita quindi viene usata in campo alimentare,
resistenza ai sali (sviluppo ad alte concentrazioni di NaCl),
fattore di adesione all'epitelio intestinale (probiotici),
resistenza all'azione di batteriofagi (preparazione di starters e sviluppo di processi fermentativi),
enzimi di restrizione (preparazione di starter e sviluppo di processi fermentativi),
formazione di idrocolloidi, ceppi filanti (effetto addensante),
enzimi per l'idrolisi dell'arginina (attività sugli aa),
sensibilità al sistema LP-SCN-H2O2 e agglutinine (controllo della crescita).
Quindi, alcuni caratteri fenotipici sono determinati da geni presenti a livello plasmidiale.
Nella cellula i plasmidi sono presenti nella forma CCC (forma circolare covalentemente chiusa) e quando si estraggono sono in questa forma, ma in alcuni casi uno dei due filamenti si rompe a dare la forma OC (circolare aperta); se si rompono entrambi i filamenti il plasmide è lineare, linear duplex (L).
Sono osservabili al microscopio a scansione.
Nella descrizione di una specie, i plasmidi non sono considerati perché possono essere facilmente persi o acquisiti dalle cellule; è comunque difficile far perdere volutamente i plasmidi alla cellula. Il curing permette ad una cellula di perdere il plasmide; serve per vedere se un'informazione è portata a livello cromosomale o plasmidiale.
Esistono due tipi di plasmidi: i primi sono implicati nei processi di trasformazione (assimilano DNA nudo) e i secondi in quelli di coniugazione (coniugativi) che si distinguono in F (fertilità) ed R (resistenza ad antibiotici *). Di alcuni plasmidi non si conoscono le funzioni, le informazioni portate, e per questo sono detti criptici.
I plasmidi si distinguono anche in base alle dimensioni.
I grandi hanno alto peso molecolare (20 milioni di Da) e sono presenti in bassa quantità nella cellula (1-5 copie); vengono trasferiti per trasformazione o coniugazione e non vengono amplificati. I piccoli hanno massimo 1000 pb, sono presenti in 10-40 copie, si trasferiscono per duplicazione cellulare e possono essere amplificati se sono compatibili con la cellula ospite (fino a 1000 copie). Questi plasmidi hanno un alto grado di amplificazione e un basso numero di cellule riceventi (es. pBr 322 di E. coli); invece i plasmidi con un basso grado di amplificazione, quelli grandi, hanno un alto numero di cellule riceventi (pRk 2PO).
I plasmidi sono spesso usati come vettori di clonaggio per produrre OGM: si taglia il loro DNA e si legano ad esso tratti di DNA differenti ottenendo un plasmide eterologo (diverso da prima), poi lo si reintroduce nel MO che diventa OGM. Una volta inserito, è importante che il vettore rimanga nelle cellule e si duplichi con esse, quindi è preferibile che il DNA plasmidiale si integri nel DNA cromosomale. Usando questo metodo si è prodotta l'insulina da E. coli (perché è un proteina piccola ed è un prodotto ad alto valore aggiunto).
*i plasmidi R posseggono anche geni che codificano per caratteristiche diverse. Ad esempio per la propria replicazione, quelli che cancellano la produzione di proteine che impediscono l'introduzione di altri plasmidi simili.
ESTRAZIONE DI PLASMIDI
Coltivazione delle cellule e lisi per estrarne il DNA (cromosomiale+plasmidi).
Denaturazione del DNA cromosomale con NaOH 3N
per 10', si riporta a pH = 7.
Il CCC non subisce l'effetto denaturante.
Ultracentrifugazione della soluzione in gradiente di cloruro di cesio e bromuro di etidio. Si preparano soluzioni di cloruro di cesio a diverse concentrazioni (gradiente di densità) e si riversano in una sola provetta dove rimangono separate a causa della loro diversa densità: quelle più leggere sopra e quelle più pesanti sotto. Dopo aver aggiunto il campione, i differenti DNA si dispongono a diversi livelli di concentrazione: il DNA cromosomale e plasmidi OC negli strati superiori perché sono più leggeri (essendo denaturati) e i plasmidi CCC più in basso perché sono più pesanti. Il bromuro di etidio, che è anche agente mutageno, si lega ai plasmidi, soprattutto in forma CCC, colorandoli di rosa-arancione; le colorazioni più marcate indicano la presenza di plasmidi. Inoltre il bromuro di etidio ha densità minore del DNA e aumenta quindi la differenza di densità tra i due tipi di DNA facilitandone la separazione. Si fotografa la lastrina. Centrifugando ottengo le due bande separate e prelevo quella sottostante con una siringa.
Elettroforesi su gel di agarosio dei plasmidi estratti e identificazione tramite confronto con DNA di plasmidi noti o tratti di DNA noto. In questo modo però si separano sul gel solo i diversi plasmidi CCC, ma non si può individuare l'eventuale presenza di un altro tipo di DNA (plasmidi OC o DNA cromosomale).
Elettroforesi bidimensionale per determinare il numero e il PM dei plasmidi e distinguere le forme OC dalle CCC ponendo la lastrina sotto una lampada UV (agente mutageno). Si taglia la lastrina e i plasmidi già separati una volta si fanno correre nell'altra direzione.
Esempio: conduco una normale elettroforesi e ottengo 3 bande.
I plasmidi sono 3 oppure 2 di cui uno presente nelle due forme (OC e CCC)?
Giro la lastra e faccio una seconda elettroforesi dopo aver denaturato con raggi UV.
La gel elettroforesi è una buona tecnica di discriminazione tra DNA cromosomale e plasmodiale. È utile su plasmidi o su frammenti di DNA cromosomale per:
determinare se il ceppo porta dei plasmidi e loro numero,
determinare dimensioni e PM in Da,
costruire una mappa di restrizione dei plasmidi, cioè si fa agire un enzima di restrizione su di essi e si determinano le funzioni che codificano (un tratto del suo DNA porta le informazioni per la propria duplicazione).
Identificare le funzioni che possono essere codificate; sono presenti le ORF, open reading frame (schema di lettura aperto), che sono frammenti di DNA, sia plasmodiale che cromosomale, che potrebbero codificare per una proteina che però non si sa quale sia.
MAPPA DI RESTRIZIONE DEI PLASMIDI
Si opera un intervento con un primo enzima di restrizione; se questo non taglia il plasmide significa che il plasmide non presenta la sequenza riconosciuta dall'enzima. Se invece una sequenza viene riconosciuta, il plasmide viene tagliato e passa alla forma OC. Un plasmide in questa forma è utile per produrre un plasmide ibrido da usare come vettore di clonaggio (meglio se produce sticky end). Se l'enzima di restrizione trova due siti di taglio si ottengono due frammenti che possono avere diverso PM e possono essere separati con elettroforesi su un gel di agarosio.
Facendo agire più enzimi di restrizione si ottengono diversi frammenti e con una gel elettroforesi si determina il loro PM; la somma dei PM delle singole frazioni rappresenta il PM del plasmide.
Oltre al PM si ricavano quali frammenti genera ogni singolo enzima e la sequenza da esso riconosciuta.
Si possono inoltre identificare le funzioni portate dai plasmidi; è stato trovato il tratto di DNA del plasmide che codifica per la capacità di autoduplicarsi (rep5) e si possono identificare gli enzimi di restrizione che tagliano questa sequenza.
Usando più enzimi diversi si riesce a ricostruire la struttura (mappa) del plasmide.
METODI DI CLONAGGIO
Il plasmide è usato come vettore di geni; si deve verificare che il plasmide rimanga nella cellula e che esprima ciò che serve, se è utile viene duplicato e inserito in MO a dare gli OGM.
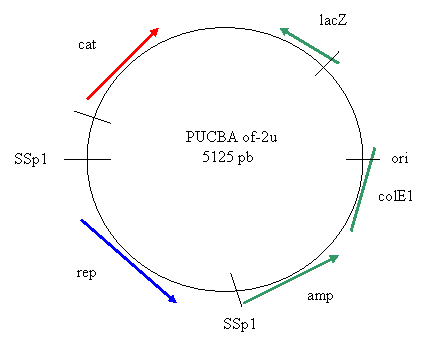
SSp1 = sono i siti di restrizione usati per costruire lo shuttle. È un plasmide shuttle perché si duplica sia in Bifidobatteri che in E. coli (se si replica in coli ha i marker verdi e in bifidi in rosso?). La parte di vettore shuttle deriva dal vettore di clonaggio commerciale pUC19 replicante in E. coli.
![]() = derivano da frammenti
ricombinanti di E. coli
= derivano da frammenti
ricombinanti di E. coli
ori = punto di origine del gene
amp = è il gene codificante per la resistenza all'ampicillina (per una lattamasi) aggiunto come marker per isolare i MO ricombinanti
colE1 = regione di amplificazione del plasmide di E. coli (presente solo in coli)
lacz = operone del lattosio, codifica per la b-galattosidasi. Usato come marker per la selezione bianco/blu, cioè il substrato cambia colore se il MO usa il galattosio
![]() cat = cloramfenicolo
acetil transferasi da St. aureus, dal plasmide pC194, usato come marker
cat = cloramfenicolo
acetil transferasi da St. aureus, dal plasmide pC194, usato come marker
![]() rep = gene codificante
per la proteina di replicazione del plasmide in bifidobatteri
rep = gene codificante
per la proteina di replicazione del plasmide in bifidobatteri
PCR (reazione a catena della polimerasi)
Quando si duplica la cellula duplica anche il proprio DNA.
La duplicazione del DNA può essere:
solo vegetativa = no coniugazione, quindi processo molto semplice e veloce
per scissione binaria.
La duplicazione (o replicazione) cellulare, o replicazione, avviene per schizogonia, cioè per scissione binaria (da un bastoncino se ne ottengono 2). Si formano tanti doppioni e non ci sono variazioni del patrimonio genetico a meno che non subentrino fattori esterni o mutazioni naturali.
La replicazione del DNA (in direzione 3'-5') è regolata e organizzata (con il DNA si duplicano i ribosomi) ed è asessuata, cioè vegetativa e quindi veloce (E. coli si duplica in 20 minuti in condizioni ottimali).
La duplicazione del DNA è semiconservativa, cioè si formano due filamenti uguali composti da un filamento vecchio e uno nuovo; questo processo non porta a variazioni del genoma a meno di mutazioni.
Inizialmente il DNA viene srotolato da una elicasi. Serve inoltre una girasi per disfare la treccia. La DNA polimerasi stende il filamento; inizia a lavorare in presenza di un doppio filamento di 10-20 pb e attacca i nucleotidi sul DNA stampo srotolato riproducendo la doppia elica (si sintetizza una catena complementare a ciascuno dei filamenti). Si ottengono due molecole di DNA, ciascuna delle quali è formata da una catena vecchia e da una nuova. Per questo motivo si parla di duplicazione semiconservativa.
Quindi per la duplicazione sono necessari: DNA a singolo filamento, un primer (tratto di DNA con funzione di innesto) e una serie di nucleotidi.
Il primer deve avere un alto %GC cosicché si leghi stabilmente al DNA. Si può usare un primer specifico, che porta una sequenza caratteristica della specie o genere del MO da ricercare (per la sua identificazione e caratterizzazione), oppure un primer con una sequenza generica. In questo caso si ottengono tanti frammenti di diverse dimensioni, mentre se è specifico si produce un solo frammento, caratteristico di quel MO. Sul frammento si potrà poi lavorare con enzimi di restrizione per determinare la sequenza nucleotidica. Le banche dati riportano le sequenze del DNA dei MO noti con cui si confronta il tratto di DNA ottenuto.
Una volta scoperto come si duplica il DNA è stato possibile mettere a punto una tecnica che consentisse l'amplificazione di segmenti di DNA in laboratorio.
È la reazione a catena della polimerasi, ovvero l'incremento (duplicazione) esponenziale in vitro di uno specifico frammento di DNA batterico.
La miscela di reazione è costituita da:
-DNA stampo (template) ss; denaturazione ottenuta aumentando la temperatura e non con elicasi. In realtà metto dentro DNA ds e poi denaturo per ottenere quello ss.
-primers, tratti di DNA di
10-20 pb. I primers devono avere un alto % GC (60-70%) per avere più legami H così
da essere stabili alle elevate temperature (90°C).
Si usano due primers o uno solo. Fungono da innesco per la DNA polimerasi. La
sequenza del primer può essere specifica o generica.
-miscela di nucleotidi
-DNA polimerasi, inizialmente si usava quella di E. coli, che però non era resistente alle alte temperature. Oggi si usa la Taq, polimerasi di Thermus aquaticus
Occorre inoltre un termociclatore, che ci permette di variare in maniera automatica le condizioni operative a livello di temperatura.
Oltre che dalla T e dal tipo di primer, la formazione dei frammenti è influenzata anche dalla concentrazione del tampone: maggiore è la concentrazione e più è stabile il legame con il template; in questo modo si amplificano solo i tratti desiderati (condizioni stringenti). Quindi si gioca su T e concentrazione per evitare che si formino frammenti senza senso.
Il ciclo di amplificazione prevede 3 fasi:
denaturazione: 95°C per 3-5''. Si ha la denaturazione del DNA ds e l'ottenimento del template (apro la doppia elica);
appaiamento (anelling): abbassamento della temperatura a circa 50°C (T<25°C di Tm) per 30-60''. Si ha l'appaiamento dei primers alla regione complementare sui singoli filamenti di DNA templato (stampo).
estensione: incremento di temperatura a 72-75°C per 30-60''. Si ha l'allungamento con sintesi di un filamento complementare al templato ad opera della polimerasi che prende i nucleotidi e li attacca.
La duplicazione avviene in direzione 3'-5', facendo riferimento al filamento che funge da stampo. Il nuovo filamento è sintetizzato in direzione da '5 a '3.
Da un filamento di DNA se ne ottengono due esattamente uguali; se si usano due primers si ottengono due filamenti vecchi interi e il resto del DNA amplificato a tratti.
Si effettuano più duplicazioni ripetendo più volte il processo, che avverrà a carico di filamenti vecchi e neoformati. Il numero di cicli che si possono ripetere varia in funzione del volume dell'apparecchio e dal risultato che vogliamo ottenere.
Conducendo da 25 a 30 cicli (2,5h se ogni ciclo dura 5 minuti) si ottengono fino a 106-107 copie dei frammenti di DNA interessato (incremento esponenziale). In realtà sono copie teoriche perché sia ha una diminuzione dell'efficienza di sintesi della DNA polimerasi per la parziale denaturazione a cui va in contro l'enzima nel susseguirsi delle fasi termiche. Oltre al danno termico, dovuto sia alla permanenza alle alte T sia alle rapide escursioni termiche tra fase di denaturazione e anelling, si deve considerare la continua variazione del pH del mezzo dovuta alla differenza di C.
Amplificazione 1-100milioni di copie
I frammenti ottenuti si separano con una elettroforesi su gel di agarosio e si leggono all'UV dopo aggiunta di bromuro di etidio.
Per vedere se il gene da amplificare codifica per l'enzima desiderato si introduce in un plasmide e poi in un MO che normalmente non ha quell'enzima (con elettroporazione); è necessario verificare che il ceppo mantenga nella cellula il plasmide e che il gene abbia un promotore forte che non si stacchi durante la trascrizione.
Enzimi di restrizione: vedo se effettivamente i segmenti hanno la stessa sequenza.
La PCR costituisce uno strumento per gestire la caratterizzazione genetico-molecolare dei MO. Posso prendere un primer che vada a pescare una sequenza specifica che ha solo il mio batterio. Così produco una sonda con la quale poter individuare il mio MO.
Se faccio una PCR generica ottengo tanti frammenti; con la sonda invece vedrò solo una banda che mi sarà data da quella sequenza specifica di DNA che ha solo quel MO.
L'estensione avviene solo quando l'estremità 3' dei primers è appaiata al templato (casi C; D ed E) In caso contrario la polimerasi non trova il substrato corretto e non svolge la sua attività (casi A e B). Inoltre maggiore è il numero di basi complementari, più stabile sarà il legame primer-stampo. Il caso E è il più favorevole all'amplificazione e darà luogo al processo anche a temperature di appaiamento più basse rispetto a C e D.
Il primer può legarsi al DNA stampo in diversi modi:
l'intero primer si lega al DNA stampo (caso ideale);
un tratto di DNA si lega e un tratto non trova la sequenza omologa, rimanendo sollevato. La sequenza non appaiata non viene amplificata; se l'estremità alzata è la 3', la sequenza non viene amplificata perché la DNA polimerasi non trova l'estremità a cui legarsi, mentre se l'estremità alzata è 5' la polimerasi può amplificare normalmente.
si legano le due estremità del primer, ma non la parte centrale; si verifica comunque l'amplificazione da parte della polimerasi.
La possibilità di avere appaiamenti parziali dei primers consente di ottenere l'amplificazione di un diverso numero di frammenti senza avere nessuna informazione sulla sequenza di quel DNA.
L'elevato %GC aumenta la stabilità degli appaiamenti parziali e l'estensione avverrà solo per quei primer che mantengono legata l'estremità 3'.
PCR: possibilità di ottenere un'impronta digitale molecolare di un MO che permette di distinguerlo da "individui" strettamente correlati dal punto di vista generico. Prima della PCR la tecnologia del fingerprinting molecolare si era basata per anni sull'analisi dei polimorfismi di lunghezza dei frammenti di restrizione, una metodica efficace ma laboriosa. La PCR è più semplice, veloce, sensibile e specifica
OPERONE RIBOSOMALE
I ribosomi sono gli organelli cellulari più numerosi, piccoli corpuscoli del citoplasma di forma quasi sferica. Sono particolarmente importanti perchè in essi avviene la sintesi proteica, ovvero la traduzione del codice genetico. Ogni proteina viene sintetizzata attraverso un processo in cui molecole di RNA messaggero escono dal nucleo e si attaccano ai ribosomi e la sequenza di RNA viene tradotta nella corrispondente sequenza di aminoacidi assemblati a formare la proteina.
Chimicamente, i ribosomi sono ribonucleoproteine, cioè strutture costituite da nucleotidi e proteine. Le grosse molecole di RNA ribosomale, rRNA (65%), formano un'impalcatura sulla quale si organizzano spontaneamente decine di tipi di proteine ribosomiche (35%).
L'RNA ribosomale è codificato da quella porzione di DNA chiamata DNA ribosomale (rDNA).
I ribosomi sono costituiti da due subunità, una maggiore e una minore, che si dissociano alla fine di ogni ciclo di sintesi di una proteina.
Esistono diversi tipi di ribosomi, questi si differenziano per la loro grandezza, che viene misurata tramite il coefficiente di sedimentazione Svedberg (S). Questa unità di misura è una costante fisica che indica la velocità con la quale i ribosomi sedimentano dopo centrifugazione in specifiche condizioni. Dipende dalle dimensioni e dalla densità della particella. Permette di distinguere ribosomi 70S e 80S.
I ribosomi 80S sono costituiti dalle subunità 40S e 60S e si trovano nel citoplasma degli eucarioti; i funghi hanno quindi ribosomi 80S. La subunità 60S è composta da rRNA di tipo 5S, 5,8S, 16-18S e 23-28S e da più di 50 proteine.
I ribosomi 70S sono tipici degli organismi procariotici e dei mitocondri e cloroplasti degli organismi eucariotici. Sono costituiti da due subunità: una con coefficiente di sedimentazione 30S (large subunit) e l'altra con coefficiente di sedimentazione 50S (small subunit). A sua volta la subunità 30S è composta da 16S rRNA e 21 proteine, mentre la subunità 50S possiede 5S, 23S rRNA e 36 proteine.
L'operone è un insieme di geni che codificano per prodotti che devono essere presenti nella cellula contemporaneamente.
È un tratto di cromosoma che contiene:
geni regolatori
siti di controllo della trascrizione
uno o più geni strutturali adiacenti trascritti in unico mRNA che codificano per le proteine di interesse
I siti di controllo sono l'operatore e il promotore.
Il promotore è la sequenza di DNA riconosciuta dalla RNApolimerasi per iniziare la trascrizione dei geni. All'interno di uno stesso DNA possono essere presenti più promotori.
L'operatore è la regione del DNA dove si lega la proteina regolatrice (repressore)
prodotta dal gene regolatore che inibisce la trascrizione dell'operone.
Ogni operone ribosomale è presente nel cromosoma batterico in numero variabile a seconda della specie.
L'operone ribosomale contiene i geni che codificano per l'RNA ribosomale ed è organizzato in geni conseguenti, legati l'uno all'altro:
-16S rDNA codifica per l'RNA 16S = 1500 pb
-SPACER che contiene il DNA che codifica per il tRNA.
-23S rDNA codifica per l'RNA 23S = 3000 pb
-5S rDNA codifica per l'RNA 5S = 100 pb
-terminatore.
16S e 23S sono regioni molto conservate, pertanto due ceppi appartengono alla stessa specie quando hanno omologia tra i diversi DNA 16 e 23S maggiore del 98% (su 1500 basi 30 sono diverse). La regione 5S è troppo piccola per dare delle informazioni.
Per determinare i rapporti filogenetici tra i MO inizialmente si confrontava l'RNA 16 S, ora si analizza anche lo spacer (piccolo) che fornisce informazioni a livello di specie e ceppo.
Un operone può essere presente nel cromosoma batterico in un numero di copie variabile a seconda della specie (in E. coli ci sono 7 copie, B. subtilis 11 copie) ed il numero di copie varia a seconda della specie.
L'operone si duplica con un meccanismo semiconservativo in direzione 3'-5', come il DNA cromosomale.
Lo spacer (regione spaziatrice) si trova tra il gene che codifica per la subunità ribosomale 16S (16S rDNA) e quello codificante la 23S (23S rDNA). Questa regione ha dimensioni (domini) diverse nelle diverse copie cromosomali dell'operone. È la parte meno conservata.
Il 16S è molto conservato quindi da questo possiamo estrarre dei primers universali. Su questo sono stati fatti degli alberi filogenetici per vedere quanto sono vicine le specie. Il 23S è già più specifico. Lo spacer dà maggiori indicazioni riguardo la specie. Lo spacer è più specifico nella caratterizzazione della specie o del ceppo.
Quindi, in base a ciò che si desidera conoscere si sceglie quale segmento amplificare.
Spesso si preferisce il 16S perché oltre ad essere molto conservato è della giusta dimensione, 1500 pb, mentre il 5S è troppo piccolo e il 23S troppo grande.
RIBOTYPING
È una metodica complessa e ormai superata. Si usano: il gene di E. coli (che codifica per l'RNA ribosomale), enzimi di restrizione e gel elettroforesi.
Serve per creare una sonda e ricercare ceppi che hanno una determinata sequenza nel loro DNA, quindi che sono della stessa specie; per vedere se un pezzo di DNA ss è complementare a un filamento ss di sequenza nota (sonda).
Il DNA batterico ribosomale è estratto e digerito in tanti piccoli frammenti con enzimi ad altra frequenza di taglio.
I frammenti vengono separati attraverso elettroforesi su gel di agarosio
Attraverso il southern blot il DNA dal gel elettroforetico viene trasferito per capillarità e fissato per adsorbimento (filtrazione e capillarità attraverso fogli di carta) su una membrana di nitrocellulosa (supporto per ibridazione).
Si ottiene il DNA ribosomale di E. coli usando la trascrittasi inversa (estratta da un fago e responsabile della conversione RNA-DNA) su RNA ribosomale del segmento 16S o 23S rRNA di E. coli. Tale DNA viene marcato con sostanze non radioattive (colorante, digossigenina) ottenendo una sonda.
Il filtro o membrana con il DNA viene posto in un tampone di ibridazione (ad alta forza ionica) contenente la sonda. Si incuba per 12-16 h a T=Tm- 3/4°C e si ha l'ibridazione tra DNA ignoto e sonda.
Lavaggio del filtro contenente gli ibridi di DNA con tampone a bassa forza ionica e ad alta temperatura, così da staccare tutti i tratti di DNA non ibridati. Condizioni di elevata stringenza. In questo modo solo la sonda complementare ibridata rimane sul supporto.
Rilevazione sonda tramite aggiunta di un anticorpo anti-digossigenina coniugato ad un gruppo rilevabile (cromogeno o fluorogeno), che si lega alla sonda. Si quantifica il DNA legato all'antibiotico (evidenziazione della posizione del legame) con una autoradiografia.
A.R.D.R.A. (amplified ribosomal DNA restriction analysis)
Analisi dei prodotti di digestione
con enzimi di restrizione
del DNA dell'operone ribosomale amplificato
È un metodo di impiego della PCR.
Viene amplificata una regione specifica e ben conservata dell'operone ribosomale, il gene che codifica per il 16S rDNA, usando un primer di E. coli.
Amplificazione dell'operone ribosomale tramite PCR (si utilizza il DNA totale senza isolare l'operone perché si impiegano primers specifici per la sequenza).
Taglio dell'operone (16S rDNA) amplificato con enzimi di restrizione specifici che riconoscono una sequenza di 4 paia di basi in modo da ottenere frammenti discreti. Ciò è necessario in quanto lavoriamo con un frammento di modeste dimensioni (1500 pb).
Separazione dei frammenti con una elettroforesi orizzontale su gel di agarosio.
Confronto dei frammenti separati con il profilo di digestione di ceppi di riferimento; due sequenze sono omologhe se hanno omologia del 98% (cioè 1470 pb su 1500 uguali). Due specie diverse dello stesso genere hanno lo stesso profilo.
L'operone ribosomiale è valido a livello del 98% di omologia genetica.
Infatti, il DNA che codifica per l'RNA 16S è talmente conservato che due ceppi appartengono alla stessa specie quando il DNA ha un grado di omologia > 98%. Se l'OG=98% potremmo essere in presenza dello stesso ceppo oppure di due ceppi diversi della stessa specie.
Bacillus cereus, antracis e thuringensis hanno omologia > al 98% (1470 pb su 1500 del 16S), ma differiscono per quanto riguarda il loro modo di essere patogeni per l'uomo. Per questo motivo i mezzi molecolari non sono sufficienti per caratterizzare un MO. (siamo a livello di genere!) Da qui la necessità di usare enzimi di restrizione che riconoscono le 4 basi in modo da ottenere un numero discreto di frammenti.
Queste tre specie oggi si raggruppano nel gruppo-specie cereus.
cereus G +, sporigeno, produttore di tossine, patogeno solo per l'uomo. Si trasmette tramite gli alimenti e dà sintomi gastrointestinali.
antracis sporigeno, tossinfezioni, armi biologiche (carbonchio, malattia che rende scuro il sangue. Contratta per inalazione del MO). Colpisce gli animali e può essere trasferito all'uomo.
thuringensis : produce una tossina ad attività insetticida nei confronti delle piralidi (ostrinia nubilaris), nel mais transgenico.
Mais BT: è mais OGM, modificato con un gene del Bacillus thuringensis. Questo Bacillus infatti produce una proteina bipiramidale che, se ingerita dalle farfalle, ne causa la morte. È attiva sulle larve di piralide, un diffuso infestante della pianta di mais. Si è isolato il gene che codifica per la proteina attiva contro le piramili e lo si è inserito nel mais.
R.A.P.D. (randomLY amplified polimorphic DNA Fingerprintings)
Impronta digitale del polimorfismo del DNA
È un metodo di impiego della PCR, che permette di ottenere un'impronta digitale che evidenzia il polimorfismo del DNA amplificato casualmente, perché casuale è la scelta dei primers, che sono aspecifici. Quindi regioni diverse di DNA cromosomale sono amplificate in modo aspecifico usando primers di sequenza arbitraria. Si ottiene un tracciato con parti di DNA con PM diverso, quindi diverse bande.
È previsto l'impiego di un solo primer contro i due necessari in una amplificazione specifica.
L'appaiamento del primer avviene in punti diversi del DNA stampo con conseguente amplificazione di un numero variabile di segmenti di diverse dimensioni. Il DNA analizzato può essere l'intero genoma (per ottenere informazioni sui geni e produrre OGM), un cromosoma o frazioni ben precise di DNA (operone ribosomale per stabilire i rapporti filogenetici tra i diversi gruppi microbici e stabilire genere e specie del MO).
È necessario separare il DNA plasmidiale per non avere interferenze.
Si fa denaturazione termica (alte temperature), poi si raffredda perché si abbia l'appaiamento col primer e si incrementa nuovamente la temperatura per consentire l'allungamento.
Si legge con una elettroforesi su gel di agarosio identificando il diverso numero e le diverse dimensioni dei frammenti ottenuti, ottenendo un profilo caratteristico di ciascun MO.
Più piccolo è il primer, più probabile è avere un gran numero di frammenti.
A partire dal DNA è possibile caratterizzare ceppi diversi appartenenti alla stessa specie e a speci diverse dello stesso genere. Questa caratterizzazione non è effettuata dal punto di vista morfologico, ma impiegando un ceppo type di riferimento.
Se è possibile conoscere l'inizio e la fine di una sequenza di DNA propria solo di una specie, è possibile produrre primers per amplificarla. Queste sequenza può essere marcata per essere visibile (reazione colorimetrica o luminosa) ed essere usata come sonda al fine di riconoscere la specie all'interno di diverse colonie. Il vantaggio della sonda è che non è più necessario isolare le colonie, ma denaturando tutte le cellule e inserendo la sonda, senza più estrarre il DNA è possibile stabilire la presenza/assenza del MO ricercato e la sua quantità. Le sonde vengono create per i MO interessanti scientificamente, soprattutto per i patogeni come Listeria.
Con l'amplificazione del DNA è possibile creare dei cluster e poi in un secondo momento caratterizzare i MO dal punto di vista morfologico per conoscerne il fenotipo.
CLUSTER: gruppo di geni codificanti per proteine con funzioni correlate (si trovano sotto il dominio di un unico operone).
I geni strutturali sono organizzati in cluster.
r.a.s. (ribosomal spacer analysis)
Analisi dello spacer
Si amplifica lo spacer, la regione spaziatrice compresa tra 16S e 23S rDNA.
È la regione meno conservata dell'operone ribosomale e quindi il polimorfismo è marcato; c'è grande variabilità per dimensione e numero da ceppo a ceppo o da specie a specie.
L'amplificazione è seguita dall'analisi dei frammenti ottenuti con una elettroforesi orizzontale su gel di agarosio.
Il primer è costituito da 20 pb posizionate circa a metà tra 16S e spacer e 23S e spacer. Sono sequenze che si riscontrano alle 2 estremità della regione spaziale, comprendenti però anche le confinanti frazioni del 16S e 23S. Quindi la regione che viene amplificata è leggermente più ampia.
Il polimorfismo della regione spaziatrice che separa i geni codificanti le subunità ribosomali 16S e 23S è maggiore perché lo spacer è la regione meno conservata, la più variabile dell'operone ribosomale per dimensione e numero di frammenti da specie a specie e da ceppo a ceppo.
Studiando lo spacer dei vari Bacillus (cereus, antracis, thuringensis e mycoides) non si riescono comunque a distinguere, anche se lo spacer è la zona più variabile in quanto l'analisi dà gli stessi risultati. Si parla dunque di specie polifasica: conoscenze fenotipiche e genotipiche che devono coincidere.
RSA nell'ambito della stessa specie: 1 banda = non c'è polimorfismo della specie; 2 o più bande c'è polimorfismo della specie. Quindi ceppi diversi.
GRUPPI MICROBICI
Dall'eubatterio ancestrale si sono sviluppati 5 gruppi microbici diversi:
cianobatteri (fotosintesi ossigenica, alghe verdi/azzure)
spirochete
batteri G +:
Clostridi
Attinomiceti
batteri purpurei
batteri sulfurei verdi
I Clostridi sono i MO più antichi e gli altri MO si sono evoluti da questi, che sono anaerobi e fermentano:
bacilli
stafilococchi
lattobacilli (i bifidobatteri non si sa dove stiano perché hanno diversi metabolismi)
omofermentanti
eterofermentanti
Leuconostoc (Oenococcus oeni)
streptococchi
Pediococchi
streptococchi lattici
enterococchi
micoplasmi
altre linee non definite (es: Enterococcus fecium e faecalis sono supposti patogeni e hanno origini più comuni rispetto a Enterococcus avium e gallinarum).
CASE STUDIES
BATTERI SPORIGENI AEROBI TERMOFILI
Sono un gruppo di Bacillus non definito filogeneticamente.
Fino al 1986 si conoscevano 3 specie sporigene anaerobie e termofile:
B. stearothermophilus, le cui spore venivano utilizzate per effettuare i controlli di sterilità in autoclave
B. coagulans, che determina la coagulazione del latte mediante acidificazione e produzione di una proteasi
B. acidocaldarius, che oggi è un Alicyclobacillus acidocaldarius
Dal 1986 al '96 sono stati proposti e validamente descritti nuovi generi e specie di batteri anaerobi; rimane comunque un gruppo non chiaro. A metà dell'800 si sosteneva il polimorfismo tra i batteri; cioè che esistessero poche specie sotto diverse forme e varietà (cocchi e bastoncini). Più avanti il concetto di specie è stato introdotto ai MO; si tratta di un concetto diverso da quello usato per gli animali superiori, per cui due organismi sono della stessa specie se hanno figli fertili.
Oggi, due MO appartengono alla stessa specie se hanno le stesse caratteristiche fenotipiche e genotipiche
È stato dunque introdotto il concetto di specie polifasica.
L'uomo ha diversi interessi verso i MO termofili, tra i quali:
capacità di alterare gli alimenti ;
patogenicità;
capacità di crescita a 37-40°C e resistenza ai principali trattamenti di risanamento degli alimenti;
produzione enzimi attivi ad alte temperature e termostabili, che sono esocellulari, quindi facili da isolare. La temperatura ottimale e la temperatura massima di crescita dei batteri termofili non hanno gli stessi valori della temperatura ottimale dei suoi enzimi isolati; l'enzima è più stabile del MO. L'enzima termostabile più usato è la TAQ polimerasi;
produzione di metaboliti di potenziale interesse applicato;
produzione di biomasse, usate come colture starter, per i processi di biotrasformazione, usati in microbiologia industriale.
Basandosi su queste considerazioni è stata effettuata una ricerca sui batteri termofili che hanno habitat in suolo, acqua, aria e sostanze organiche in decomposizione. I termofili del suolo si trovano in micronicchie anaerobiche.
Vediamo le diverse fasi del lavoro di ricerca.
1- CARATTERISTICHE FENOTIPICHE
Ottenimento dei ceppi: estratti dal suolo di diverse aree geografiche (14) e da collezioni (ceppi type) usati per confrontare i ceppi del suolo.
Pastorizzazione: 1g di sospensione di suolo è stata sospesa in 5g (mL) di acqua distillata sterile (o tampone) e pastorizzata a 90°C per 10'.
Isolamento e coltura: i MO dei campioni sono stati isolati per diluizioni successive e seminati in un terreno colturale agarizzato (nutrient agar, contenente estratto di carne, peptone e sali). Le piastre sono incubate a 55°C per 6-12h. Non si usano temperature superiori per evitare che l'acqua del terreno evapori concentrando i nutrienti. Con questo metodo si possono perdere dei MO perché la temperatura di incubazione e il terreno utilizzati sono selettivi; si perdono MO mesofili, sporigeni non sporificati e la pastorizzazione può aver eliminato tutte le forme vegetative di una specie (tindalizzazione, la pastorizzazione facilita la sporulazione). L'isolamento è ripetuto più volte per ottenere colture pure.
Conservazione: le piastre vengono mantenute in frigo a +4°C. Se si usano in tempi lunghi, una volta si conservavano a -80°C in brodo comune con 15% di glicerolo (evita che si formino grossi cristalli di ghiaccio dentro e fuori che rompono le cellule); oggi si liofilizzano le cellule in un ambiente che protegga.
Dall'analisi al microscopio delle colture isolate si è visto che:
sono tutti bastoncellari, singoli o in catenelle;. dimensioni non definite perché presenti molte specie diverse
sporigeni, ma non definite le singole caratteristiche (non esistono cocchi sporigeni)
aerobi
alcuni possono cambiare Gram (detti Gram variabili perché hanno diverse pareti)
termofili perché crescono tutti a 55 e 65°C. Sono stati ottenuti 85 isolati (non si sa se della stessa specie o ceppo)
2- CARATTERISTICHE GENETICO-MOLECOLARI
I ceppi analizzati sono:
85 di nostro isolamento
17 type o di riferimento
Da ognuno di questi è stato estratto il DNA e usate la tecniche:
ARDRA
ARDRA-ITS (internal trancripted spacer) che è un abbinamento tra i due metodi e l'oggetto dell'analisi è lo spacer (ITS)
RAPD, con primer universali (codificano per tutto il DNA, non solo per il 16S) o mirati; si usa il primer OPI-07 (Operon Technologies Inc), con alto %GC, di 10 pb (sequenza: 5'-CAGCGACAAG-3').
RSA (ribosomal spacer amplification) analisi dello spacer.
Nell'ARDRA è stato analizzato il 16S, amplificato con la PCR e tagliato con diversi enzimi di restrizione. Il risultato non è condizionato dai primers, perché sono universali, ma dall'enzima di restrizione. Nell'ARDRA-ITS si ricerca il frammento composto da 16S + ITS + 1840 pb del 23S; in totale circa 2000 pb. Gli enzimi di restrizione usati sono:
H ae III
T aq I
A lu I
S au 3A
che tagliano in diversi punti dando diversi profili elettroforetici, ma riconoscono tutti una sequenza di 4 pb in modo da ottenere frammenti piccoli (una sequenza più lunga trova meno frammenti ma più lunghi); i primer usati sono specifici.
Nell'ARDRA sono stati analizzati 20 ceppi a caso (random) ed è stato scelto l'Hae III per confrontare i diversi profili elettroforetici.
Gli isolati e i type sono stati riuniti in gruppi di affinità in base all'attività di questo enzima. In alcuni casi all'interno dei gruppi c'è l'83% di omologia genetica; in altri casi il 3-56%.
I° gruppo: 43 isolati e 7 type.
Quindi è relativamente omogeneo perché contiene molti type (magari tutti uguali).
I 7 type sono:
![]() B. caldotenax
B. caldotenax
B. caldovelox
B. caldolyticus magari tutte specie vere
B. kaustophilus
![]() B. thermoleovorans
B. thermoleovorans
B. stearothermophilus DSH2 magari ceppi diversi
B. stearothermophilus ATCC
II° gruppo: 34 isolati e 1 type (B. thermodenitrificans).
III° gruppo: 3 isolati e 1 type (B. thermoglucosidasius).
IV° gruppo: 1 isolato con profilo uguale a due type (B. sphaercus e B. thermosphaericus) che magari sono due ceppi della stessa specie, in quanto differiscono solo per la diversa temperatura.
V°gruppo: 2 isolati e 1 type (B. pallidus).
VI° gruppo: 2 isolati, che non hanno profilo simile, ma il loro profilo non è simile a nessun type, neanche al B. flavothermus.
Rimangono 2 type: B. thermocatenolatum e i 2 stearothermophilus ATCC type e non type, diversi da tutti i MO analizzati.
L'ARDRA sul 16S ha i seguenti svantaggi: il frammento analizzato ha molta omologia tra i ceppi e l'analisi è influenzata dal tipo di enzima di restrizione usato (con AluI però sono stati confermati i risultati ottenuti con HaeIII, solo con qualche differenza tra i type).
Si conclude che c'è molta eterogeneità tra i ceppi (profili elettroforetici diversi da tutti gli altri)
L'ARDRA-ITS conferma i risultati già ottenuti, quindi sono conservati sia il 16S che il 23S.
La RAPD raggruppa gli 85 isolati e i 17 type in 15 profili elettroforetici distinti, ottenendo gli stessi risultati per gli isolati.
L'RSA usando l'enzima Tab II genera ancora i risultati della RAPD.
CONCLUSIONI
Degli 85 isolati, abbiamo 80 ceppi che sono:
43 B. thermoleovorans
34 B. thermodenitrificans
2 B. pallidus
1 B. thermosphaericus
Questi costituiscono il 94,11% degli isolati che siamo riusciti ad inquadrare in generi o speci abbastanza sicuri.
Gli altri 5 ceppi, 5.89%, potrebbero essere generi o specie nuovi.
Esiste una eterogeneità genotipica nella specie B. thermoleovorans e B. stearothermophilus. (o aereot?)
Depositati i ceppi type se trovati generi e specie differenti.
È possibile con la genetica molecolare abbinata all'omologia DNA/DNA identificare circa il 90% dei ceppi sporigeni aerobi termofili.
RICADUTE
ridefinizione della specie B. thermodenitrificans
creato un nuovo genere e una nuova specie: Ureibacillus terrenus
riclassificato il Saccarococcus caldo-xylosi-lyticus in Geobacillus caldo-xylosi-lyticus, MO oggetto di 3 pubblicazioni i riviste internazionali.
PEDIOCOCCUS PENTOSACEUS E ACIDILATTICI
I pediococchi acidilacti e pentosaceus sono batteri lattici di grande interesse per il settore agroalimentare. Sono omofermentanti e si trovano nei vegetali fermentati quali olive, cetrioli, crauti e nel settore lattiero caseario come popolazione microbica secondaria. Diversi ceppi di Pediococcus acidilattici sono stati isolati da: carne, orzo, patate.
Hanno 2 aspetti positivi:
usati come colture starter nei vegetali e negli insaccati
usati come biopreservanti perché, essendo acidificanti, abbassano il pH. Inoltre producono delle batteriocine, le pediocine, che impediscono lo sviluppo di L. monocitogenes, S. aureus e C. botulinum
Sono due specie non distinguibili in base al fenotipo.
È possibile distinguere le due specie attraverso il molecolare. Si tratta infatti di specie diverse, e forse addirittura di generi diversi. La differenza di specie è data dall'analisi del genoma (%GC e % di omologia genetico).
Il %GC di P. acidilattici (Pa) è 41-44% mentre quello di P. pentosaceus (Pp) è di 37-39%, quindi sono specie distinguibili. La % di omologia genetica tra Pa e Pp è del 40% quindi sono due specie diverse. Invece, l'analisi del DNA cromosomale non dà alcuna informazione, neanche l'analisi dello spacer.
Il segmento utilizzato per riconoscere le due specie è un gene coinvolto nella sintesi della lattodeidrogenasi L e D. Inoltre nelle due specie, soprattutto nel Pa, sono stati individuati dei ceppi produttori di una particolare pediocina (AcH- PA-1, il gene produttore è il ped Af); da questa si risale alla sequenza nucleotidica e ai primer.
Si confronta la sequenza di questi geni con banche dati. Ma questa tecnica è complessa e richiede molto tempo e una grande attenzione. Pertanto, essendo due MO di interesse industriale, si è rivelato necessario trovare un metodo più semplice per differenziarli.
I geni codificanti per la lattodeidrogenasi dei due MO erano già stati scoperti e depositati, pertanto è stato sufficiente effettuare una ricerca in banca dati attraverso cui si è compreso che ogni specie ha un gene specifico, diverso dall'altro. A questo punto è stato costruito il primer sulla parte con meno omologia rispetto alla sequenza del gene del secondo MO. Si amplifica il DNA con metodo ARDRA e ne risulta, a seguito della corsa elettroforetica, una banda definita per ogni specie. È quindi stato possibile costruire due sonde.
Attraverso la PCR è possibile, inoltre, effettuare una distinzione nell'ambito della stessa specie. Si può infatti stabilire quali MO sono Pediococcus acidilacti e quali fra di essi sono anche produttori di pediocine.
[Il gene che codifica per la lattodeidrogenasi è house-keeping, ovvero costruttivo. Al contrario, il gene che codifica per la pediocina è inducibile]
Come per la lattodeidrogenasi, in banca dati viene ricercata la sequenza genica per la pediocina. Sono stati creati i primers ed è stata effettuata l'amplificazione.
1-PCR MULTIPLA/MULTILOCI
Si conduce una PCR usando i primer per la LDH e per la batteriocina e si distinguono le diverse specie e ceppi e tra gli stessi ceppi quali producono al batteriocina.
 1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8
Il frammento a maggior PM è il gene per le LDH (caratteristico del P. acidilattici) mentre il frammento a minor PM è il prodotto di amplificazione del gene che codifica per la pediocina. I ceppi 2-3-4 sono P. acidilattici che producono la pediocina; mentre 6-7-8 non producono la batteriocina.
2- AGAR GERMI: PROVA DI CRESCITA/INIBIZIONE
Si valuta l'attività antibiotica.
Lactobacillus plantarum è sensibile alla batteriocina; viene inoculato in un terreno di coltura adatto al suo sviluppo cosicché il MO cresca creando una patina uniforme sulla superficie (inoculo consistente in modo che occupi tutta la piastra). Poi si appoggiano sul terreno dei dischetti di carta da filtro imbevuti di filtrato colturale di pediococchi, contenente la batteriocina, diluiti o di diversi ceppi. Non è un'analisi totalmente quantitativa. Si mette la piastra in frigo per 10h (overnight) per permettere alla pediocina di diffondere nel terreno colturale e impedire al L. plantarum di crescere. Si incuba la piastra e si ottiene crescita uniforme con degli aloni di inibizione in prossimità dei dischetti contenenti la pediocina, se questa era presente nel liquido colturale.
[stessa cosa al contrario se il dischetto è imbevuto di vitamine indispensabili per la crescita microbica: alone di crescita]
3- SOUTHERN BLOT
Per verificare la presenza di pediocina.
I frammenti di DNA dei pediococchi produttori di pediocine vengono dapprima trasferiti dal gel a una membrana di nitrocellulosa o matrici simili (nylon) e denaturati. La membrana viene poi messa in contatto con una sonda marcata per poter permettere la formazione di ibridi nel caso in cui nella sonda vi siano sequenze complementari a uno dei frammenti, i quali sono fissati sulla membrana e mantenuti in condizioni denaturanti perché non possano rigenerare molecole a doppia elica. I siti di ibridazione possono essere riconosciuti osservando se la sonda marcata si è "legata" o meno alla membrana. Questo procedimento viene effettuato come controllo della produzione di pediocine da parte del MO.
(Dot blot è procedura semplificata di southern blot per il riconoscimento dei MO).
TOMA PIEMONTESE DOP
È un prodotto tradizionale ottenuto da latte crudo messo a riposo per 12h a 8-10 °C in assenza di starter.
Poi viene posto in caldaia con il caglio per 40-60' a 37-40 °C, a temperatura, tempo e pH non controllati. La cagliata viene estratta e rotta in frammenti di 1 cm (per 10-15'), messa in fascere per darle la forma, pressata per eliminare il siero e asciugata per 24h. La stagionatura dura 30-40 d a 6-10 °C e con umidità pari all'85% in grotte o caverne.
Utilizzando latte crudo sono presenti dei MO sconosciuti. Lo scopo dello studio è caratterizzare le popolazioni microbiche dominanti responsabili delle caratteristiche della toma (non starter).
È stata effettuata una caratterizzazione del prodotto dal punto di vista microbiologico, tecnologico e sensoriale. Sono stati analizzati i MO che caratterizzano un prodotto tipico attraverso l'isolamento di 116 forme di cocchi e utilizzando tutte le applicazioni molecolari: analisi dello spacer RSA, ricercate sonde specifiche ARDRA, sequenza di un frammento di 500 pb del DNA che codifica per l'RNA 16S (usato per aspetti filogenetici).
Popolazioni microbiche:
70 % Lattococchi (predominano i cocchi), crescono a 10 °C (maturazione) e in presenza di 6% di NaCl.
la forma prevalente è il Lactococcus lactis, che presenta due subspecie:
- Lactococcus lactis lactis è un gruppo-specie complesso che comprende più specie
- Lactococcus lactis cremoris dà aroma di burro alla panna, non lo si ritrova in natura ma solo nei caseifici
rilevante il Lactococcus gravieae (patogeno dei pesci, non è detto che si patogeno per l'uomo). Quello dei formaggi è uguale al patogeno dei pesci (problema tassonomico e diagnostico per individuarli) si trova nel latte mastitico, ma non è agente patogeno di tale malattia.
Entrambi i MO sono potenziali patogeni e sono simili tra loro.
È stata prodotta una toma esente da Lactococcus gravieae, ma si è ottenuto un formaggio diverso, quindi questo MO è necessario per la produzione di toma.
Bisogna stabilire se il MO è patogeno.
Ma come?
Si determina la sequenza aminoacidica dell'enzima (i patogeni producono enzimi)
Si ricava il gene che codifica per l'enzima
Si ricerca in primer in banca dati
Si introduce il gene in un plasmide o nel DNA cromosomale (Corynebacterium difterie)
Si fa il curigo e si vede se il MO lo produce ancora
Si separano i due DNA (plasmidiale e cromosomale) e si ricerca la sequenza (i plasmidi possono diventare lineari e confondersi col DNA cromosomale)
Come E. coli ha ceppi non patogeni e il ceppo O 157: H 7. Si isola il MO dal formaggio e si confronta con ceppi patogeni. Si può introdurre il gene che codifica per la patogenicità in plasmidi e iniettare il MO in topi; il topo può non morire nel caso in cui il gene non si sia espresso totalmente, è necessario usare promotori forti.
Per verificare il tipo di MO si verifica il postulato di Kock.
Tra i cocchi è stata trovata una specie nuova: Enterococcus italicus
Sono stati eliminati alcuni generi, il Saccharococcus è un bastoncellare
BACILLUS LICHENIFORMIS
Le proteasi alcaline termostabili sono enzimi proteolitici termostabili che lavorano a pH alcalini, usati nella formulazione di biodetersivi.
Tra i MO più idonei per la produzione di tali enzimi esocellulari ci sono quelli del genere Bacillus (G+, aerobi o anaerobi facoltativi) tra cui il B. subtilis e licheniformis.
Nel licheniformis sono state trovate delle endonucleasi di restrizione e dei plasmidi nuovi.
La specie è determinata in termine polifasico, cioè ha caratteristiche fenotipiche e genotipiche ben definite; è un MO sporigeno (il vantaggio di lavorare con gli sporigeni è che le spore resistono a lungo) ha aspetto di lichene, crostoso, alcune volte è colorato di rosa. Effettua una respirazione aerobia dove può usare propinato come unica fonte di carbonio e una anaerobia in cui usa i nitrati. Queste caratteristiche permettono di isolare quasi esclusivamente il B. licheniformis
È stata fatta un'analisi su 182 campioni di terra provenienti da tutto il mondo. Il campione di terra è stato trattato termicamente a 180°C per eliminare tutte le forme vegetative e il MO è stato isolato in condizioni aerobie in presenza di propinato. (l'unica fonte di carbonio che è in grado di utilizzare)
MORFOLOGIA
Il licheniformis analizzato ha la stessa morfologia degli altri Bacillus:
Gram +
bastoncellare, 0.6-0.8 m (diametro) per 1.5-3.0 m (l), a volte in catenelle
mobile per flagelli laterali
l'endospora è ovale o cilindrica in posizione centrale o subterminale
lo sporangio non si deforma visibilmente (non è più gonfio della forma vegetativa)
FONTI DI CARBONIO (sistema API)
È presente un certo grado di biodiversità nella specie:
il 100% utilizza le stesse 21 fonti di C
il 100% non è in grado di usare altre 17 fonti di C
altre 12 fonti di C sono usate in maniera differente dai diversi ceppi
altre 50 fonti di C sono usate in maniera differente
CRESCITA IN NaCl
Ancora alta biodiversità, alcuni MO sono abbastanza alofili
il 100% cresce al 6-8% di NaCl
il 30% cresce al 12% di NaCl
il 5% cresce al 15% di NaCl
TEMPERATURA
quasi un vero termofilo (quasi perché in grado di crescere anche a 27°C)
il 100% cresce a 55°C
il 70% cresce a 60°C
il 55% cresce a 65°C
PLASMIDI
Solo il 22% ha dei plasmidi, alcuni ne hanno 3 per cellula, altri uno solo. Il loro PM va da >30pb a 2.6 pb.
%GC:
45.7-49.8, quindi c'è variazione tra i ceppi analizzati
46.7 è %GC del type del B. licheniformis
batteriocine
Il 70% risulta positivo alla produzione di batteriocine
OMOLOGIA GENETICA
Dei 182 ceppi analizzati:
161 hanno omologia da 66 a 100%, quindi sono uguali tra loro (A)
tra i restanti 21 ceppi si distinguono due gruppi, che tra loro hanno bassa omologia (3-37%):
16 ceppi che tra loro hanno l'84% di omologia genetica (B)
i restanti 5 ceppi che tra loro hanno l'84% di omologia (C).
I gruppi B e C sono stati confrontati con altre specie di Bacillus: pumilis, subtilis, cereus, coagulans, ma l'omologia genetica con questi è bassa (20%) quindi non appartengono a queste specie. I gruppi B e C appartengono alla specie licheniformis perché hanno le sue stesse caratteristiche fenotipiche, ma sono considerati delle variazioni della specie (varianti genotipiche) chiamati genomovar.
Esistono anche le biovar = varianti biologiche, e le sierovar = varianti sierotipiche.
Sui gruppi B e C sono state condotte analisi con diversi metodi molecolari (ARDRA e RAPD) ma gli enzimi di restrizione non permettono di ottenere alcuna informazione che differenzi tra loro le genomovar; ha quindi più valore l'omologia genetica che non consente però di distinguere le specie. È stata usata una sonda caratteristica del licheniformis (gene cgtA) ma non è stata utile perché altri geni hanno la stessa omologia di questo.
Sono stati studiati due plasmidi criptici di B. licheniformis (pFL5) di cui è stata analizzata la sequenza nucleotidica e l'organizzazione strutturale, cioè la sequenza di geni o dei possibili geni del plasmide (open reading frame).
Il problema è tuttora aperto
IL MOLECOLARE
Gli utilizzi del molecolare sono molteplici:
diagnostica: risolto il problema dei due Pediococchi (acidilacti e pentosaceus), fatte sonde per individuare i MO patogeni, evidenziato un nuovo problema di doppia specie (B. cereus)
biodiversità: evidenziate le differenze intraspecie, trovati generi e specie nuovi e sono state create le collezioni, cioè banche per la conservazione del germoplasma batterico.
qualità microbiologica: di prodotti fermentati; un prodotto è tale perché ha certi MO. Importante soprattutto nei prodotti tipici (es. toma)
tradizioni alimentari confermate: se cambiano le formulazioni cambiano i prodotti. Es vino: attualmente prodotto in tank di acciaio inox. Per conferirgli le caratteristiche sensoriali che acquisiva dal legno delle botti, vengono aggiunti dei trucioli.
carte di identità: salvaguardano gli alimenti e soprattutto i prodotti tradizionali.
tipicità alimentare: salvaguarda gli alimenti.
Alcuni vogliono eliminare i prodotti tipici perché pensano che siano ricchi di MO patogeni; i MO possono dare problemi agli organismi non abituati alla loro presenza perché sono abituati ad alimenti pastorizzati o sterilizzati.
L'obiettivo del microbiologo è prevenire che l'alimento si contamini di MO alterativi o patogeni, quindi assicurare la sicurezza d'uso. Studiando i seguenti aspetti si sono trovate specie nuove e più biotipi appartenenti alla stessa specie.
Il microbiologo alimentare deve dunque assicurare i seguenti aspetti:
prevenzione e sicurezza: la prevenzione e sicurezza microbiologica degli alimenti (non contaminazione chimica o biologica) riguarda la microbiologia predittiva (HACCP, analisi dei rischi e dei punti critici). Esempio: Legionella negli impianti di condizionamento. Per individuare i MO patogeni (E. coli, Yersinia, Salmonella (kit), Listeria (kit)) si amplificano determinati geni con la PCR e si fanno sonde nucleiche.
qualità microbiologica degli alimenti: la qualità microbiologica stabilisce che un alimento ha certi tipi di MO, compresi i MO non starter. Questi MO producono batteriocine (nisina) e/o abbassano il pH dell'alimento conferendo le caratteristiche sensoriali al prodotto ed esplicando anche una funzione protettiva.
tipicità: La tipicità microbiologica dei prodotti è data dalla presenza di alcuni MO caratteristici di quei prodotti. È importante la salvaguardia delle tradizioni dei nostri alimenti. Un prodotto tradizionale ha un elevato contenuto in MO con molta biodiversità, non ancora totalmente nota.
Già nell'antichità le attività metaboliche dei MO venivano sfruttate per la produzione di alimenti.
Gli egiziani producevano la birra da orzo maltato. All'epoca di Mosè si producevano latti fermentati, vino e pane lievitato.
I processi industriali non usano MO naturalmente presenti sull'alimento, ma degli starter su prodotto pastorizzato perché hanno meno scarti di prodotto, che risulta uniforme e di qualità costante, ma con appiattimento del gusto.
Nello yogurt si usano ceppi omofermentanti, Lactobacillus bulgaricus e Streptococcus lactis, per cui il 90% dello zucchero viene convertito in acido lattico. Questi MO producono anche diacetile e acetaldeide.
Nel kefir intervengono batteri lattici e lieviti.
Nel burro oltre ai MO che producono acidità(L. lactis e cremoris) sono presenti ma anche quelli che formano diacetile e acetoina; (L. lactis, L. diacetilactis, Leuconostoc citrovorum e dextranicum).
Nei formaggi si ha produzione di acidi più lenta in modo da favorire l'attività del caglio, lo spurgo ed evitare lo sviluppo di altri MO. I MO starter poi muoiono e i loro enzimi rimangono attivi sulle componenti del formaggio durante la stagionatura. Nel grana intervengono Lactobacillus helveticus, bulgaricus e casei. Nell'Emmental sono presenti anche batteri proponici (anche nel Montasio), che usano i prodotti dei batteri lattici ed hanno lipasi e proteasi che creano un prodotto aromatico e saporito.
OGM: ORGANISMO GENETICAMENTE MODIFICATO
Il primo vettore su cui è stata dimostrata l'efficacia è costituito dal vettore plasmide Ti, tumour inducine, presente dell'Agrobacterium tumefaciens che causa tumori nelle piante (es. grano). Questo plasmide ha la caratteristica di integrarsi efficacemente nei cromosomi della pianta causandone il tumore.
Dal plasmide Ti sono stati eliminati i geni responsabili della patogenicità senza alterare l'aggressività nei confronti delle piante. Al loro posto è stato inserito il gene che dà la capacità di fissare N atmosferic. Ma questo progetto non è stato portato avanti per non alterare il mercato dei concimi.
La preoccupazione relativa alla produzione di OGM è legata a due possibili eventi:
possibile propagazione dei transgenici, cioè dei nuovi caratteri ad altri organismi con alterazione degli equilibri ambientali (possibile inquinamento da DNA nell'ambiente e conseguente fenomeno di resistenza.
Eventuale pericolosità di alimenti di origine vegetale e animale ottenuti da piante e animali transgenici (allergenicità)
Un esempio attuale di vegetale transgenico è il mais Bt, che contiene il gene del Bacillus thuringensis codificante per un corpo parasporale proteico che ha la forma di un doppio prisma ed ha la capacità di forare lo stomaco delle larve di Ostrinia nubilaris. Dato che questa piralide attacca la pianta a livello di foglie e fusto, è stato fatto in modo che l'espresione della proteina avvenisse a livello di tali zone. Se viene prodotto mais Bt è necessario che la zona coltivata sia affiancata da un campo incolto che faccia da rifugio per insetti scoraggiaiti all'attacco del mais, ma ciò crea problemi di spazio.
La "Monsanto" è il lider mondiale degli ibridi. Esisteva un diserbante totale creato da questa azienda a cui resisteva soltanto una pianta, sempre prodotta dalla stessa azienda.
Poi si è passati al mais bt e si è instaurato un monopolio totale. In seguito sempre la monsanto creò il mais terminator: ibrido resistente, ma il seme non in grado di germinare e si era costretti a riacquistare le sementi. Ribellinone.
In Europa è vietato l'uso di mais e soia transgenici.
Nel 2006 è stata consentita la presenza di OGM non mischiati con normali alimenti.
Gli alimenti possono contenere massimo 0.9% di OGM (non si può fare di meglio per limite degli strumenti) e tale presenza deve essere riportata in etichetta. La presenza di OGM va dichiarata però solo se gli OGM sono presenti nella formulazione dell'alimento, ma non se l'animale è stato nutrito con alimenti transgenici.
Pane e pasta: negli USA c'è rischio basso di trovare OGM perché non è consentita la produzione e l'uso di frumento transgenico (regola non valida per la produzione di alimenti per celiaci). Problema: aggiunte al normale pane comune, ad esempio a base di soia.
Riso: nei paesi poveri c'è il problema della cecità infantile, dovuta a carenza di b-carotene. È stato dunque creato il golden rice, riso a cui viene aggiunto il gene per sintetizzare b-carotene. Può anche essere alimento per celiaci.
Carne e pesce: basso rischio di presenza di OGM perché gli animali non si cibano di alimenti transgenici, eccetto i pesci e i polli da allevamento per cui si usano mangimi di soia. (salmoni OGM di dimensioni doppie).
Frutta e verdura: rischio medio-basso; sono transgenici: papaia, pomodori, zucchine e barbabietola da zucchero. In Europa non è consentita la vendita di OGM mentre in USA si. Nel coltivare ortofrutta transgenico è opportuno controllare che non si verifichino fenomeni di resistenza in altri prodotti.
Cibi pronti: rischio alto perché molti contengono soia o suoi derivati transgenici, tipo lecitina.. (il 60% della soia coltivata è OGM).
Caffè: quello transgenico contiene meno caffeina e permette di uniformare il tempo di maturazione e raccolta, con riduzione dei costi.
Vino: con lieviti transgenici per accelerare il processo di vinificazione.
Bibite e succhi di frutta: da mais OGM (maltosio).
Uova: da galline alimentate con OGM.
Merendine: ad alto rischio perché prodotte con mais dolce (transgenico). È difficile scoprire OGM presenti nel prodotto finito.
Il rischi di mangiare un alimento contenente ingredienti transgenici è maggiore nei prodotti manipolati.
Ci sono batteri transgenici usati in campo industriale e diagnostico, muffe in campo industriale.
Batteri lattici alcuni utilizzati come tali; è stato trovato lo Streptococcus mutans che è responsabile della carie perché produce batteriocine, il suo mutante è stato modificato in modo che non produca acido lattico.
Pseudomonas iceminus mutato non produce delle proteine che sono nucleo di aggregazione di ghiaccio e causano le gelate tardive.
S. cerevisiae mutato conduce la fermentazione malolattica, usato da australiani non usato per i vini perché diminuisce l'acidità.
SEMINARIO FORTINA
Studio su Lactobacillus helveticus, MO termofilo omofermentente, importante nell'ambito del settore lattiero caseario, perché fa parte del sieroinnesto per la produzione di formaggi di alta qualità, come Grana e Provolone.
In precedenza sono stati fatti pochi studi genetici in qunto questo MO è diverso dagli altri lattobacilli termofili (es. L. bulgaricus) per la sua organizzazione genica, quindi non era possibile utilizzare gli stessi modelli.
Analizzano il suo fenotipo è possibile ottenere molte informazioni. Alcuni ceppi appartenenti alla stessa specie sono dotati di una maggiore attività acidificante o di una attività proteolitica più pronunciata, o ancora ad una maggior resistenza al lisozima rispetto ad altri.
Pertanto è possibile selezionare non più di una specie, ma un ceppo starter appartenente alla specie, che presenti le caratteristiche ottimali in funzione del prodotto che si vuole ottenere (ad es. posso scegliere un ceppo ad elevata attività acidificante e bassa attività proteolitica, o viceversa).
L'elevata biodiversità fenotipica dipende dal genotipo, motivo per il quale è stata condotta un'indagine sulla biodiversità genotipioca, in particolare sulla presenza di geni legati ad attività particolari, fino ad allora poco conosciuti.
I ceppi di Lactobacillus helveticus sono stati isolati da lattoinnesti. Per questo MO non era ancora conosciuto l'operone del lattodeidrogenasi e la sua organizzazone, ma lo studio di questo insieme di geni è importante al fine di conoscere in che modo il lattosio viene acquisito e acidificato.
Gli ORF sono regioni codificanti delle quali però non si conosce l'espressione. Sono gruppi contigui di codoni nucleotidici non sovrapposti in una molecola di DNA o RNA, comprese le regioni di termine. Si trasformano in geni, cioè sequenze polinucleotidiche con sito di inizio (ATG, GTG, TTG o CTG) e di fine.
I geni sono formati da due siti principali:
Sito promotore a monte; è il sito che permette il legame dell'ER al sito dell'mRNA, che poi indurrà la trascrizione.
Sito operatore è addetto alla regolazione dell'espressione del gene.
I alcuni casi i geni attigui vengono regolati ed espressi insieme, al fine di ottenere un risparmio energetico per la cellula. Si ha quindi un operone, insieme di geni che codificano per prodotti che devono essere presenti nella cellula contemporaneamente.
L'operone è caratterizzato dall'avere un solo promotore e un solo operatore a monte del primo dell'insieme di geni.
Il promotore è costituito da due sequenze consenso:
Tata box: TATAAT, in cui le prime tre basi devono essere conservate, quindi deve essere possibile trovarle in una sequenza non nota, in cui le ultime tre basi possono variare. È anche detta sito-10 perché teoricamente questa sequenza è a 10 basi dall'inizio del gene.
Sito-35: TTGACA. Anche in questo caso le prime tre basi sono conservate e le ultime tre variabili. È un sito importante perché come il tata box viene riconosciuto dalla RNApolimerasi.
Tanto più queste due sequenze sono conservate, tanto più il promotore è forte e affine alla DNA polimerasi, quindi maggiore sarà la velocità di trascrizione.
Questo è un sistema della cellula per regolare i propri geni.
In realtà non è semplice individuare il promotore ed è importante sapere se è forte o debole, quindi quanto velocemente codifica e produce l'mRNA per il metabolita che ci interessa. Conoscendo il promotore è poi anche possibile, nel caso sia debole, sostituirlo con uno forte e ottenere così un gene migliorato.
Si individua poi il sito RBS, sito di legame al ribosoma, situato 6-7 basi prima del codone d'inizio ATG, si trova in una regione in cui guanina e adenina sono le basi più rappresentate.
La RNA polimerasi si attacca al filamento inserendosi solo nel punto in cui riconosce le sequenze consenso; l'enzima è infatti composto da due subunità che riconoscono la presenza delle sequenze -35 e -10, prima del gene da codificare.
La velocità con cui l'enzima si lega al filamento di DNA è maggiore se le due sequenze corrispondenti ai due siti sono conservate (TATAAT/TTGACA). Questo legame forte consente la traduzione dell'mRNA. Queste due sequenze possono subire delle variazioni: tanto meno forte sarà il legame del DNA con la RNA polimerasi, tanto più lenta sarà la trascrizione.
Vicino al sito promotore, c'è il sito operatore, in grado di formare un legame con una proteina che può fungere da repressore o attivatore; quindi l'operatore agisce in sinergia con il promotore al fine di regolare l'espressione genica.
Il gene regolatore produce una proteina che ha affinità di legame con il sito operatore e può essere un repressore o un attivatore.
se è un repressore, legandosi all'operatore impedisce il legame della RNA polimerasi al filamento di DNA sul sito promotore e di conseguenza blocca la sintesi dell'mRNA e del metabolita corrispondente al gene non trascritto.
se è un attivatore, si legherà al promotore in modo che questo divenga forte e permetta la trascrizione immediata dell'mRNA e la sintesi del metabolita. L'attivatore non agisce modificando la sequenza nucleotidica dei due siti -10 e -35, ma sposta la RNA polimerasi in modo che i siti siano maggiormente disponibili. Infatti, oltre alla composizione delle sequenze, è importante la distanza, che dovrebbe essere di 16-17 basi.
Per il Lactobacillus helveticus sono stati evidenziati due meccanismi per prelevare lattosio: un sistema fototransferasico e uno presamico.
Vediamo meglio il sistema presamico.
Vi è una proteina di trasporto, la permeasi, una proteina di membrana che riconosce la presenza di lattosio all'esterno della cellula, lo lega e lo trasporta all'interno con un dispendio energetico. Dentro la cellula il lattosio viene scisso in glucosio e galattosio dalla b-galattosidasi. In seguito un altro enzima preleva il galattosio e ne permette l'impiego attraverso la via glicolitica trasformandolo in un intermedio.
Per il Lactobacillus helveticus non si conosceva nulla, pertanto si pensò che avesse la stessa organizzazione degli altri lattobacilli: permeasi, b-galattosio e il terzo enzima per trasformare il lattosio. Tuttavia nelle banche dati non era depositata la sequenza genica del batterio.
Si voleva trovare un enzima presente con la stessa funzione in tutti i batteri lattici: gli enzimi con la stessa attività catalitica avranno nella sequenza amminoacidica, quindi anche in quella nucleotidica, alcune zone conservate. In particolar modo quelle corrispondenti al sito catalitico, in quanto questo deve svolgere la stessa funzione nell'ambito di tutti gli enzimi simili, quindi sarà codificato dalla stessa sequenza.
Si vuole quindi ricercare la sequenza di un gene housekeeping non ancora noto per una data specie batterica.
La strategia da adottare è la seguente:
cercare in banca dati sequenze note codificanti per la b-galattosidasi, presenti in specie correlate e non.
allineare le sequenze per compararle e ricercare una regione con bassissime differenze fra le basi, quindi conservare fra tutte le sequenze in esame, in modo tale che possano avere corrispondenze con domini amminoacidici, come il dominio catalitico.
disegnare primers idonei agli estremi della regione da amplificare (il forward va esattamente come si lega, il reverse, invece, va indicato come complementare della lettura).
allestire amplificazioni specifiche: se il risultato è positivo si determina la sequenza della posizione amplificata per la specie in esame e poi si può effettuare il sequenziamento.
Individuata la coppia di primers (15pb) si estrae il DNA e si fa la PCR.
Si prende un frammento del gene amplificato e si fa l'elettroforesi. In seguito lo si sequenzia grazie ad un sequenziatore (ad ogni colore e picco corrisponde una base nucleotidica).
In tal modo si ottiene una sequenza parziale del gene codificante per la b-galattosidasi del Lactobacillus helveticus. Ma si vuole ancora sapere se tale gene appartiene ad un operone.
Si mette allora a punto una PCR con primers divergenti.
Si induce La DNA polimerasi a sintetizzare frammenti di DNA al di fuori di quello già ottenuto; ciò può essere fatto perché a questo punto si ha a disposizione la sequenza specifica. È un sistema semplice ma lungo, perché va fatto per passaggi successivi: ogni volta va sequenziato il frammento finchè no si trovano i codoni di inizio e fine.
In seguito si va al sito di inizio per verificare la presenza di promotore, operatore, TATA box e sito-35, al fine di poter giustificare la sequenza di un gene attraverso una attività pro
Sono stati trovati:
TTGTTT sito -35
TATAAT sito-10
GAGG RBS
Con ATG si ha l'inizio della regione codificante per la b-galattosidasi. In questo caso si tratta di un gene e non di un ORF.
Sapendo che la b-galattosidasi è inserita nel promotore, è stato trovato un gruppo di geni (cluster) uguali a quelli dei lattobacilli. Si è in presenza di un operone associato a geni singoli, che partecipano insieme all'acquisizione e scissione del lattosio e alla trasformazione del galattosio.
La b-galattosidasi è codificata da due geni sovrapposti (perchè letti in frame diversi) lacL e lacM; sono 2 ORF sovrapposte che rappresentano due subunità di una b-galattosidasi caratteristica di Lactobacillus helveticus e di qualche altro lattobacillo.
È stato effettuato il sequenziamento di due geni vicini e la sequenza è stata comparata in banca dati per capire la loro funzione attraverso similitudini con altre sequenze già depositate.
Nel L. helveticus questi due geni sovrapposti (lacL e lacM) hanno azione combinata al fine di dare un unico prodotto (b-galattosidasi). A monte di lacL e lacM è stato individuato il promotore, ma dopo questi due geni è stato trovato un ORF successivo: galE, ovvero galattosio epimerasi.
Il sito terminatore fra lacM e galE è molto debole: in particolari condizioni la cellula è in grado di sintetizzare i 3 geni assieme, quindi siamo in presenza di un operone.
Il galE è costitutivamente trascritto dal MO.
Al contrario lacL e lacM sono inducibili: l'ultilizzo del lattosio da parte del L. helveticus è inducibile; in presenza di lattosio l'unico promotore che funziona è quello a monte dei 3 geni, quindi vengono sintetizzati assieme.
Se il lattosio non è presente (es. coltivo su ribosio) lacL e lacM sono repressi, mentre galE, che comunque ha un promotore proprio, può essere trascritto.
Quello di galE è quindi un gene costitutivo che può essere espresso da solo, ma è correlato a due geni inducibili. Il gene regolatore lacR è un repressore, infatti blocca lacM e lacL in assenza di lattosio. Quando però il lattosio è presente è in grado di staccare la proteina del sito repressore e quindi attivare la trascrizione di b-galattosidasi.
Nel L. helveticus, sul filamento opposto, in direzione inversa a quella dei geni per la b-galattosidasi, è sono stati trovati due geni per la permeasi contigui (lacS e lacS') di cui uno non è attivo.
|
| Appunti su: |
|